
MicroRNAs and Growth Factors: An Alliance Propelling Tumor Progression

Abstract
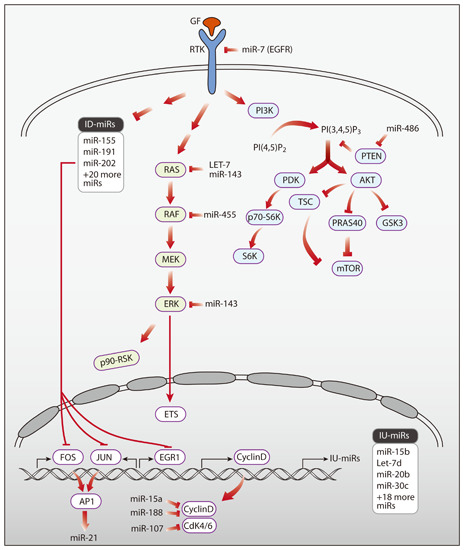
:
1. Introduction

2. Occurrence and Biogenesis of microRNAs and Their Relevance to Cancer

3. Networks of Growth Factors and microRNAs
3.1. Growth Factors Regulating miRNAs

3.2. Specific microRNAs Regulating Growth Factor Signaling
3.3. Feedback Loops Linking microRNAs and Growth Factors
4. Potential Clinical Applications of miRNAs Relevant to Growth Factors and Signal Transduction
5. MicroRNAs as Molecular Targets of Future Cancer Therapeutics
6. MicroRNAs as Modulators of Patient Response to Drugs Targeting Growth Factor Signaling

{kind=link}
{kind=link}
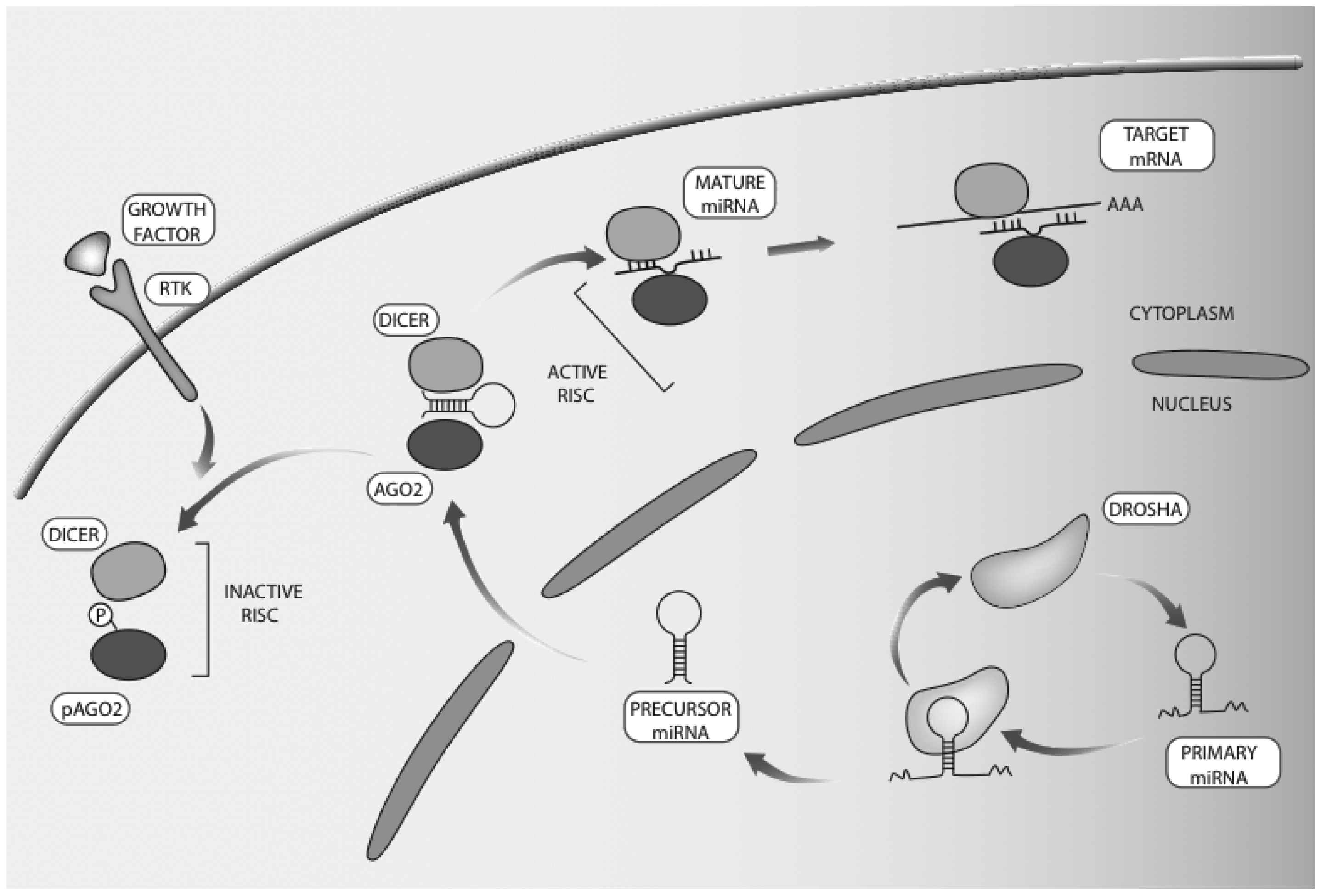{kind=link}
{kind=link}
| miRNA | Target Gene(s) | Effect | Drug & Tumor | Reference |
|---|---|---|---|---|
| miR-566 | VHL | Knockdown of miR-566 inhibited cell proliferation and invasion and led to cell cycle arrest in glioma cells. It further sensitized glioblastoma cells to Nimotuzumab | Nimotuzumab (glioblastoma) | [119] |
| miR-200c | ZNF217, ZEB1 | Overexpression of miR-200c increased sensitivity to trastuzumab and suppressed invasiveness of breast cancer cell lines | Trastuzumab (breast) | [116] |
| miR-375 | IGF1R | Overexpression of miR-375 restored sensitivity to trastuzumab resistant cell lines and increased the efficacy of trastuzumab in a xenogarft model. | Trastuzumab (breast) | [115] |
| miR-221 | PTEN | Overexpression of miR-221 inhibited apoptosis, promoted metastasis and induced trastuzumab resistance in HER-2 positive breast cancer cells. | Trastuzumab (breast) | [120] |
| miR-200c | ERRFI-1 | Overexpression of miR-200c regains sensitivity of the resistant cell lines to cetuximab treatment resulting in reduced cell growth in vitro | Cetuximab (bladder) | [117] |
| miR-146a | EGFR signaling | Overexpression of miR-146a suppressed cell growth and increased cellular apoptosis in HCC cell lines and displayed synergistic effects with cetuximab | Cetuximab (hepatocellular) | [118] |
| miR-7 | EGFR | Overexpression of miR-7 enhanced the effect of erlotinib on growth inhibition of FaDu cells | Erlotinib (head&neck) | [114] |
| miRNA | Target Gene(s) | Effect | Drug & Tumor | Reference |
|---|---|---|---|---|
| miR-30b/30c and miR-221/222 | BIM, APAF-1 (respectively) | Knockdown of miR-30b, -30c, -221 and -222 in gefitinib-resistant cells induces increased sensitivity gefitinib | Gefitinib (lung) | [121] |
| miR-21 | PTEN | Overexpression of miR-21 decreased sensitivity of lung cells to gefitinib. Knock-down of miR-21 restored gefitinib sensitivity of the corresponding gefitinib-resistant cell line and caused a dramatic reduction in tumor size | Gefitinib (lung) | [122] |
| miR-34a | MET | Overexpression of miR-34a in EGFR mutant NSCLC increased sensitivity to gefitinib, resulting in increased inhibition of cell growth and to induced apoptosis, which resulted in tumor regression | Gefitinib (lung) | [124] |
| miR-138-5p | GPR124 | Overexpression of miR-138-5p in NSCLC cells increased sensitivity to gefitinib in vitro | Gefitinib (lung) | [123] |
| miR-103 and miR-203 | PKC-e, SRC (respectively) | Overexpression of miR-103 and miR-203 increased sensitivity to gefitinib in lung cells resistant to the drug | Gefitinib (lung) | [121] |
| miR-203 | EREG, TGFA, API5, BIRC2, TRIAP1 | Overexpression of miR-203 synergistically enhanced the effect of CI-1033 on reduction of tumor size in a xenograft model of nude mice injected with Ras-activated cells | CI-1033 (prostate) | [125] |
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
Author Contributions
References
- Solomon, E.; Borrow, J.; Goddard, A.D. Chromosome aberrations and cancer. Science 1991, 254, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. How does multistep tumorigenesis really proceed? Cancer Discov. 2015, 5, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Antal, T.; Dingli, D.; Traulsen, A.; Kinzler, K.W.; Velculescu, V.E.; Vogelstein, B.; Nowak, M.A. Genetic progression and the waiting time to cancer. PLoS Comput. Biol. 2007, 3, e225. [Google Scholar] [CrossRef] [PubMed]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010, 25, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Todaro, G.J. Autocrine secretion and malignant transformation of cells. N. Engl. J. Med. 1980, 303, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Fusetti, L.; Villa, R.; Sozzi, G.; Minoletti, F.; Mauri, P.; Pierotti, M.A. Transforming activity of the chimeric sequence formed by the fusion of collagen gene col1a1 and the platelet derived growth factor b-chain gene in dermatofibrosarcoma protuberans. Oncogene 1998, 17, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, A.; O’Brien, K.P.; Sjoblom, T.; Pietras, K.; Buchdunger, E.; Collins, V.P.; Heldin, C.H.; Dumanski, J.P.; Ostman, A. The dermatofibrosarcoma protuberans-associated collagen type ialpha1/platelet-derived growth factor (pdgf) b-chain fusion gene generates a transforming protein that is processed to functional pdgf-bb. Cancer Res. 1999, 59, 3719–3723. [Google Scholar] [PubMed]
- Hynes, N.E.; Watson, C.J. Mammary gland growth factors: Roles in normal development and in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003186. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [PubMed]
- Tarcic, G.; Avraham, R.; Pines, G.; Amit, I.; Shay, T.; Lu, Y.; Zwang, Y.; Katz, M.; Ben-Chetrit, N.; Jacob-Hirsch, J.; et al. Egr1 and the erk-erf axis drive mammary cell migration in response to egf. FASEB J. 2012, 26, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic foxo transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J.; et al. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 2007, 39, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Tullai, J.W.; Schaffer, M.E.; Mullenbrock, S.; Sholder, G.; Kasif, S.; Cooper, G.M. Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J. Biol. Chem. 2007, 282, 23981–23995. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of egfr signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The drosha-dgcr8 complex in primary microrna processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and mechanisms related to rna interference regulate expression of the small temporal rnas that control c. Elegans developmental timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Song, J.J.; Smith, S.K.; Hannon, G.J.; Joshua-Tor, L. Crystal structure of argonaute and its implications for risc slicer activity. Science 2004, 305, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Finn, K.J.; Ji, X.; Baillat, D.; Gregory, R.I.; Liebhaber, S.A.; Pasquinelli, A.E.; Shiekhattar, R. Microrna silencing through risc recruitment of eif6. Nature 2007, 447, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Tijsterman, M.; Plasterk, R.H. Dicers at risc; the mechanism of rnai. Cell 2004, 117, 1–3. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microrna targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D.; Haley, B. Ribo-gnome: The big world of small rnas. Science 2005, 309, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of micrornas on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M.; Calin, G.A. Mirnas, cancer, and stem cell division. Cell 2005, 122, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. Microrna biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Lieberman, J. Dysregulation of microrna biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Braye, S.G.; Forbes, J.F.; Scott, R.J. The expression of dicer and drosha in matched normal tissues, tumours and lymph node metastases in triple negative breast cancer. BMC Cancer 2014, 14, 253. [Google Scholar] [CrossRef] [PubMed]
- Poursadegh Zonouzi, A.A.; Nejatizadeh, A.; Rahmati-Yamchi, M.; Fardmanesh, H.; Shakerizadeh, S.; Poursadegh Zonouzi, A.; Nejati-Koshki, K.; Shekari, M. Dysregulated expression of dicer in invasive ductal breast carcinoma. Med. Oncol. 2015, 32, 643. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, B.; Iuliani, M.; Zoccoli, A.; Pantano, F.; Fioramonti, M.; de Lisi, D.; Frezza, A.M.; Rabitti, C.; Perrone, G.; Onetti Muda, A.; et al. Deregulation of dicer and mir-155 expression in liposarcoma. Oncotarget 2015, 6, 10586–10591. [Google Scholar] [PubMed]
- Shen, J.; Xia, W.; Khotskaya, Y.B.; Huo, L.; Nakanishi, K.; Lim, S.O.; Du, Y.; Wang, Y.; Chang, W.C.; Chen, C.H.; et al. Egfr modulates microrna maturation in response to hypoxia through phosphorylation of ago2. Nature 2013, 497, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. Smad proteins control drosha-mediated microrna maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Sas-Chen, A.; Manor, O.; Steinfeld, I.; Shalgi, R.; Tarcic, G.; Bossel, N.; Zeisel, A.; Amit, I.; Zwang, Y.; et al. Egf decreases the abundance of micrornas that restrain oncogenic transcription factors. Sci. Signal. 2010, 3, ra43. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, M.; Ben-Chetrit, N.; Korner, C.; Mancini, M.; Ben-Moshe, N.B.; Lauriola, M.; Lavi, S.; Biagioni, F.; Carvalho, S.; Cohen-Dvashi, H.; et al. Egf induces micrornas that target suppressors of cell migration: Mir-15b targets mtss1 in breast cancer. Sci. Signal. 2015, 8, ra29. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Amit, I.; Citri, A.; Shay, T.; Carvalho, S.; Lavi, S.; Milanezi, F.; Lyass, L.; Amariglio, N.; Jacob-Hirsch, J.; et al. A reciprocal tensin-3-cten switch mediates egf-driven mammary cell migration. Nat. Cell Biol. 2007, 9, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Hummel, M.; Pantano, L.; Pastor, X.; Vivancos, A.; Castillo, E.; Mattlin, H.; Ferrer, A.; Ingham, M.; Noguera, M.; et al. Microarray and deep sequencing cross-platform analysis of the mirrnome and isomir variation in response to epidermal growth factor. BMC Genomics 2013, 14, 371. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.C.; Kao, S.Y.; Yang, C.C.; Tu, H.F.; Wu, C.H.; Chang, K.W.; Lin, S.C. Egf up-regulates mir-31 through the c/ebpbeta signal cascade in oral carcinoma. PLoS ONE 2014, 9, e108049. [Google Scholar] [CrossRef] [PubMed]
- Ben-Chetrit, N.; Chetrit, D.; Russell, R.; Korner, C.; Mancini, M.; Abdul-Hai, A.; Itkin, T.; Carvalho, S.; Cohen-Dvashi, H.; Koestler, W.J.; et al. Synaptojanin 2 is a druggable mediator of metastasis and the gene is overexpressed and amplified in breast cancer. Sci. Signal. 2015, 8, ra7. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, I.; Hoffmann, P.; Adelson, D.L. Micrornas are part of the regulatory network that controls egf induced apoptosis, including elements of the jak/stat pathway, in a431 cells. PLoS ONE 2015, 10, e0120337. [Google Scholar] [CrossRef] [PubMed]
- Gomez, G.G.; Volinia, S.; Croce, C.M.; Zanca, C.; Li, M.; Emnett, R.; Gutmann, D.H.; Brennan, C.W.; Furnari, F.B.; Cavenee, W.K. Suppression of microrna-9 by mutant egfr signaling upregulates foxp1 to enhance glioblastoma tumorigenicity. Cancer Res. 2014, 74, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.J.; Martiniez, A.; Shi, X.B.; Yang, J.; Evans, C.P.; Dobi, A.; deVere White, R.W.; Kung, H.J. Mir-30 as a tumor suppressor connects egf/src signal to erg and emt. Oncogene 2014, 33, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. Microrna-7 inhibits the epidermal growth factor receptor and the akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.J.; Giles, K.M.; Price, K.J.; Zhang, P.M.; Mattick, J.S.; Leedman, P.J. Regulation of epidermal growth factor receptor signaling in human cancer cells by microrna-7. J. Biol. Chem. 2009, 284, 5731–5741. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Bemis, L.T.; Nakajima, E.; Sugita, M.; Birks, D.K.; Robinson, W.A.; Varella-Garcia, M.; Bunn, P.A., Jr.; Haney, J.; Helfrich, B.A.; et al. Egfr regulation by microrna in lung cancer: Correlation with clinical response and survival to gefitinib and egfr expression in cell lines. Ann. Oncol.: Off. J. Eur. Soc. Med. Oncol./ESMO 2008, 19, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Chiyomaru, T.; Seki, N.; Inoguchi, S.; Ishihara, T.; Mataki, H.; Matsushita, R.; Goto, Y.; Nishikawa, R.; Tatarano, S.; Itesako, T.; et al. Dual regulation of receptor tyrosine kinase genes egfr and c-met by the tumor-suppressive microrna-23b/27b cluster in bladder cancer. Int. J. Oncol. 2015, 46, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Hsiao, T.H.; Hong, T.M.; Chen, H.Y.; Kao, S.H.; Wang, W.L.; Yu, S.L.; Lin, C.W.; Yang, P.C. Microrna-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PLoS ONE 2014, 9, e96765. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Zhang, S.; Shan, C.; Zhou, L.; Zhou, Z. Microrna-133a regulates the cell cycle and proliferation of breast cancer cells by targeting epidermal growth factor receptor through the egfr/akt signaling pathway. FEBS J. 2013, 280, 3962–3974. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shao, X.; Gao, W.; Zhang, Z.; Liu, P.; Wang, R.; Huang, P.; Yin, Y.; Shu, Y. Microrna-133b inhibits the growth of non-small-cell lung cancer by targeting the epidermal growth factor receptor. FEBS J. 2012, 279, 3800–3812. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, E.; Wendt, K.L.; Augustine, L.A.; Stecklein, S.R.; Sibala, E.C.; Li, D.; Gunewardena, S.; Jensen, R.A. Brca1 regulation of epidermal growth factor receptor (egfr) expression in human breast cancer cells involves microrna-146a and is critical for its tumor suppressor function. Oncogene 2014. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Zheng, X.; Jiang, F.; Rogers, T.; Szalad, A.; Chopp, M. Mir-146b-5p suppresses egfr expression and reduces in vitro migration and invasion of glioma. Cancer Investig. 2010, 28, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.A.; Arimappamagan, A.; Pandey, P.; Santosh, V.; Hegde, A.S.; Chandramouli, B.A.; Somasundaram, K. Mir-219-5p inhibits receptor tyrosine kinase pathway by targeting egfr in glioblastoma. PLoS ONE 2013, 8, e63164. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yao, J.; Shi, X.; Hu, L.; Li, Z.; Song, T.; Huang, C. Microrna-302b suppresses cell proliferation by targeting egfr in human hepatocellular carcinoma smmc-7721 cells. BMC Cancer 2013, 13, 448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schiff, D.; Park, D.; Abounader, R. Microrna-608 and microrna-34a regulate chordoma malignancy by targeting egfr, bcl-xl and met. PLoS ONE 2014, 9, e91546. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, S.; Mannsperger, H.; Zhang, J.D.; Horvat, E.A.; Schmidt, C.; Kublbeck, M.; Henjes, F.; Ward, A.; Tschulena, U.; Zweig, K.; et al. Global microrna level regulation of egfr-driven cell-cycle protein network in breast cancer. Mol. Syst. Biol. 2012, 8, 570. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, A.; Valvo, C.; Fabrizi, E.; di Martino, S.; Biffoni, M.; Runci, D.; Forte, S.; de Maria, R.; Ricci-Vitiani, L. Analysis of the combined action of mir-143 and mir-145 on oncogenic pathways in colorectal cancer cells reveals a coordinate program of gene repression. Oncogene 2013, 32, 4806–4813. [Google Scholar] [CrossRef] [PubMed]
- Pekow, J.R.; Dougherty, U.; Mustafi, R.; Zhu, H.; Kocherginsky, M.; Rubin, D.T.; Hanauer, S.B.; Hart, J.; Chang, E.B.; Fichera, A.; et al. Mir-143 and mir-145 are downregulated in ulcerative colitis: Putative regulators of inflammation and protooncogenes. Inflamm. Bowel Dis. 2012, 18, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Niu, X.; Zhang, X.; Tao, J.; Wu, D.; Wang, Z.; Li, P.; Zhang, W.; Wu, H.; Feng, N.; et al. Mir-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of kras. Mol. Cell. Biochem. 2011, 350, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bhayani, M.K.; Dodge, C.T.; Nicoloso, M.S.; Chen, Y.; Yan, X.; Adachi, M.; Thomas, L.; Galer, C.E.; Jiffar, T.; et al. Coordinated targeting of the egfr signaling axis by microrna-27a*. Oncotarget 2013, 4, 1388–1398. [Google Scholar] [PubMed]
- Leivonen, S.K.; Sahlberg, K.K.; Makela, R.; Due, E.U.; Kallioniemi, O.; Borresen-Dale, A.L.; Perala, M. High-throughput screens identify micrornas essential for her2 positive breast cancer cell growth. Mol. Oncol. 2014, 8, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.M.; Barker, A.; Zhang, P.M.; Epis, M.R.; Leedman, P.J. Microrna regulation of growth factor receptor signaling in human cancer cells. Methods Mol. Biol. 2011, 676, 147–163. [Google Scholar] [PubMed]
- Epis, M.R.; Giles, K.M.; Barker, A.; Kendrick, T.S.; Leedman, P.J. Mir-331-3p regulates erbb-2 expression and androgen receptor signaling in prostate cancer. J. Biol. Chem. 2009, 284, 24696–24704. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, A.; Bayerlova, M.; Strotbek, M.; Schmid, S.; Beissbarth, T.; Olayioye, M.A. A global microrna screen identifies regulators of the erbb receptor signaling network. Cell Commun. Signal. 2015, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Goga, A.; Bhaumik, D.; Berger, C.E.; Sullivan, C.S.; Benz, C.C. Coordinate suppression of erbb2 and erbb3 by enforced expression of micro-rna mir-125a or mir-125b. J. Biol. Chem. 2007, 282, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Liu, M.; Yan, X.; Zhou, Y.; Wang, W.; Wang, X.; Fu, Z.; Wang, N.; Zhang, S.; Wang, Y.; et al. Mir-193a-3p functions as a tumor suppressor in lung cancer by down-regulating erbb4. J. Biol. Chem. 2015, 290, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, J.; Yan, M.; Liu, L.; Lin, H.; Zhao, F.; Sun, L.; Zhang, Y.; Cui, Y.; Zhang, F.; et al. Microrna-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the erbb4/pik3r3/mtor/s6k2 signaling pathway. Oncogene 2015, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, Q.; Zhang, L.; Zhou, S.; Ye, W.; Yao, Q.; Li, Z.; Huang, C.; Wen, Q.; Wang, J. Mir-302b is a potential molecular marker of esophageal squamous cell carcinoma and functions as a tumor suppressor by targeting erbb4. J. Exp. Clin. Cancer Res. 2014, 33, 10. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Chow, A.S.; Au, J.S. Mir-145 inhibits cell proliferation of human lung adenocarcinoma by targeting egfr and nudt1. RNA Biol. 2011, 8, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Dougherty, U.; Robinson, V.; Mustafi, R.; Pekow, J.; Kupfer, S.; Li, Y.C.; Hart, J.; Goss, K.; Fichera, A.; et al. Egfr signals downregulate tumor suppressors mir-143 and mir-145 in western diet-promoted murine colon cancer: Role of g1 regulators. Mol. Cancer Res. 2011, 9, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Zhang, C.; Shi, J.; Xu, M.H.; Liu, F.; Yuan, H.H.; Wang, J.Y.; Jiang, B.; Gao, F.H. Abnormal activation of the egfr signaling pathway mediates the downregulation of mir145 through the erk1/2 in non-small cell lung cancer. Oncol. Rep. 2014, 31, 1940–1946. [Google Scholar] [PubMed]
- Zhang, K.L.; Han, L.; Chen, L.Y.; Shi, Z.D.; Yang, M.; Ren, Y.; Chen, L.C.; Zhang, J.X.; Pu, P.Y.; Kang, C.S. Blockage of a mir-21/egfr regulatory feedback loop augments anti-egfr therapy in glioblastomas. Cancer Lett. 2014, 342, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Seike, M.; Goto, A.; Okano, T.; Bowman, E.D.; Schetter, A.J.; Horikawa, I.; Mathe, E.A.; Jen, J.; Yang, P.; Sugimura, H.; et al. Mir-21 is an egfr-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- rna genes mir15 and mir16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating micrornas as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of micrornas in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 micrornas in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Baldwin, D.A.; Scearce, L.M.; Oberholtzer, J.C.; Tobias, J.W.; Mourelatos, Z. Microarray-based, high-throughput gene expression profiling of micrornas. Nat. Methods 2004, 1, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific micrornas from mouse. Curr. Biol.: CB 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Rosenfeld, N.; Aharonov, R.; Meiri, E.; Rosenwald, S.; Spector, Y.; Zepeniuk, M.; Benjamin, H.; Shabes, N.; Tabak, S.; Levy, A.; et al. Micrornas accurately identify cancer tissue origin. Nat. Biotechnol. 2008, 26, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Git, A.; Graf, S.; Salmon-Divon, M.; Curtis, C.; Sottoriva, A.; Zhao, Y.; Hirst, M.; Armisen, J.; Miska, E.A.; et al. The shaping and functional consequences of the microrna landscape in breast cancer. Nature 2013, 497, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. Microrna expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C.A.; Necela, B.M.; Thompson, E.A.; Perez, E.A. Microrna signatures: Clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Enerly, E.; Steinfeld, I.; Kleivi, K.; Leivonen, S.K.; Aure, M.R.; Russnes, H.G.; Ronneberg, J.A.; Johnsen, H.; Navon, R.; Rodland, E.; et al. Mirna-mrna integrated analysis reveals roles for mirnas in primary breast tumors. PLoS ONE 2011, 6, e16915. [Google Scholar] [CrossRef] [PubMed]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microrna molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, X.; Zhao, Y.; Tian, T.; Jin, G.; Shu, Y.; Chen, Y.; Xu, L.; Zen, K.; Zhang, C.; et al. Serum microrna signatures identified in a genome-wide serum microrna expression profiling predict survival of non-small-cell lung cancer. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C. Micrornas: Potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int. J. Biochem. Cell Biol. 2010, 42, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Meiri, E.; Mueller, W.C.; Rosenwald, S.; Zepeniuk, M.; Klinke, E.; Edmonston, T.B.; Werner, M.; Lass, U.; Barshack, I.; Feinmesser, M.; et al. A second-generation microrna-based assay for diagnosing tumor tissue origin. Oncologist 2012, 17, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.S.; Maeda, L.S.; Ioannidis, J.P. Clinical outcome prediction by micrornas in human cancer: A systematic review. J. Natl. Cancer Inst. 2012, 104, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting micrornas in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Shibata, C.; Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Ohno, M.; Takata, A.; Koike, K. Current status of mirna-targeting therapeutics and preclinical studies against gastroenterological carcinoma. Mol. Cell. Ther. 2013, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of micrornas in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. Lna-mediated microrna silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ameres, S.L.; Friedline, R.; Hung, J.H.; Zhang, Y.; Xie, Q.; Zhong, L.; Su, Q.; He, R.; Li, M.; et al. Long-term, efficient inhibition of microrna function in mice using raav vectors. Nat. Methods 2012, 9, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. Microrna silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. Microrna sponges: Competitive inhibitors of small rnas in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.Y.; Giraldez, A.J.; Schier, A.F. Target protectors reveal dampening and balancing of nodal agonist and antagonist by mir-430. Science 2007, 318, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microrna mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther.: J. Am. Soc. Gene Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microrna delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Pfeffer, S.; Baumert, T.F. Mir-122 acts as a tumor suppressor in hepatocarcinogenesis in vivo. J. Hepatol. 2013, 58, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis c virus rna abundance by a liver-specific microrna. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of hcv infection by targeting microrna. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microrna component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microrna mir-34a inhibits prostate cancer stem cells and metastasis by directly repressing cd44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.L.; Jiang, Q.Y.; Jin, X.; Shen, J.; Wang, K.; Li, Y.B.; Xu, F.J.; Tang, G.P.; Li, Z.H. Cationic microrna-delivering nanovectors with bifunctional peptides for efficient treatment of panc-1 xenograft model. Biomaterials 2013, 34, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.; Vallely, M.P.; et al. Restoring expression of mir-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol.: Off. J. Eur. Soc. Med. Oncol./ESMO 2013, 24, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The erbb network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Shaw, A.T. Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Mori, F.; Biagioni, F.; Bellissimo, T.; Pulito, C.; Muti, P.; Strano, S.; Blandino, G. Micrornas: Short non-coding players in cancer chemoresistance. Mol. Cell. Ther. 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Hummel, R.; Hussey, D.J.; Haier, J. Micrornas: Predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur. J. Cancer 2010, 46, 298–311. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.G.; Finn, S.P.; Cuffe, S.; O’Byrne, K.J.; Barr, M.P. The emerging role of micrornas in resistance to lung cancer treatments. Cancer Treat. Rev. 2015, 41, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, F.C.; Giles, K.M.; Candy, P.A.; Ali, A.; Ganda, C.; Epis, M.R.; Webster, R.J.; Leedman, P.J. Regulation of epidermal growth factor receptor signaling and erlotinib sensitivity in head and neck cancer cells by mir-7. PLoS ONE 2012, 7, e47067. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.M.; Zhu, H.Y.; Bai, W.D.; Wang, T.; Wang, L.; Chen, Y.; Yang, A.G.; Jia, L.T. Epigenetic silencing of mir-375 induces trastuzumab resistance in her2-positive breast cancer by targeting igf1r. BMC Cancer 2014, 14, 134. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.D.; Ye, X.M.; Zhang, M.Y.; Zhu, H.Y.; Xi, W.J.; Huang, X.; Zhao, J.; Gu, B.; Zheng, G.X.; Yang, A.G.; et al. Mir-200c suppresses tgf-beta signaling and counteracts trastuzumab resistance and metastasis by targeting znf217 and zeb1 in breast cancer. Int. J. Cancer. J. Int. Cancer 2014, 135, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Adam, L.; Zhong, M.; Choi, W.; Qi, W.; Nicoloso, M.; Arora, A.; Calin, G.; Wang, H.; Siefker-Radtke, A.; McConkey, D.; et al. Mir-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2009, 15, 5060–5072. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; He, R.; Rong, M.; Dang, Y.; Chen, G. Synergistic effect of mir-146a mimic and cetuximab on hepatocellular carcinoma cells. BioMed Res. Int. 2014, 2014, 384121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.L.; Zhou, X.; Han, L.; Chen, L.Y.; Chen, L.C.; Shi, Z.D.; Yang, M.; Ren, Y.; Yang, J.X.; Frank, T.S.; et al. Microrna-566 activates egfr signaling and its inhibition sensitizes glioblastoma cells to nimotuzumab. Mol. Cancer 2014, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Bai, W.; Zhu, H.; Zhang, X.; Chen, Y.; Wang, L.; Yang, A.; Zhao, J.; Jia, L. Mir-221 promotes trastuzumab-resistance and metastasis in her2-positive breast cancers by targeting pten. BMB Rep. 2014, 47, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Romano, G.; di Leva, G.; Nuovo, G.; Jeon, Y.J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. Egfr and met receptor tyrosine kinase-altered microrna expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2012, 18, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhu, F.; Liu, J.; Xu, T.; Pei, D.; Wang, R.; Qian, Y.; Li, Q.; Wang, L.; Shi, Z.; et al. Alteration in mir-21/pten expression modulates gefitinib resistance in non-small cell lung cancer. PLoS ONE 2014, 9, e103305. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, X.; Li, W.; Ping, W.; Deng, Y.; Fu, X. Mir-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating g protein-coupled receptor 124. Biochem. Biophys. Res. Commun. 2014, 446, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Chen, X.; Zhao, J.; Bao, Z.; Chen, X.; Zhang, P.; Liu, Z.F.; Zhou, J.Y. Microrna-34a overcomes hgf-mediated gefitinib resistance in egfr mutant lung cancer cells partly by targeting met. Cancer Lett. 2014, 351, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.K.; Abou-Kheir, W.; Yin, J.J.; Chang, Y.S.; Barrett, B.; Suau, F.; Casey, O.; Chen, W.Y.; Fang, L.; Hynes, P.; et al. Loss of egfr signaling regulated mir-203 promotes prostate cancer bone metastasis and tyrosine kinase inhibitors resistance. Oncotarget 2014, 5, 3770–3784. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kedmi, M.; Sas-Chen, A.; Yarden, Y. MicroRNAs and Growth Factors: An Alliance Propelling Tumor Progression. J. Clin. Med. 2015, 4, 1578-1599. https://doi.org/10.3390/jcm4081578
Kedmi M, Sas-Chen A, Yarden Y. MicroRNAs and Growth Factors: An Alliance Propelling Tumor Progression. Journal of Clinical Medicine. 2015; 4(8):1578-1599. https://doi.org/10.3390/jcm4081578
Chicago/Turabian StyleKedmi, Merav, Aldema Sas-Chen, and Yosef Yarden. 2015. "MicroRNAs and Growth Factors: An Alliance Propelling Tumor Progression" Journal of Clinical Medicine 4, no. 8: 1578-1599. https://doi.org/10.3390/jcm4081578
APA StyleKedmi, M., Sas-Chen, A., & Yarden, Y. (2015). MicroRNAs and Growth Factors: An Alliance Propelling Tumor Progression. Journal of Clinical Medicine, 4(8), 1578-1599. https://doi.org/10.3390/jcm4081578