
Reversible Human TGF-? Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Signaling Pathways Controlling Liver EMT
2.1. Hepatic EMT
2.2. Involvement of Both Pro-Inflammatory Cytokines and TGF-β Signals via JNK in Hepatic EMT
3. Multiple Phospho-Isoforms of Smad2 and Smad3 Exist
3.1. TGF-β Signaling
3.2. JNK Signaling
3.3. Smad Phospho-Isoform Signaling
4. Involvement of Smad Singaling in EMT
4.1. Mitogenic pSmad3L (Ser-213) Pathway
4.2. Fibrogenic TβRI/JNK/pSmad2L (Ser-245/250/255)/C Signaling
4.3. Immature pSmad2/3L (Thr-220/179)/C Signaling
5. Similarities and Differences of the TGF-β Signal between Epithelial and Mesenchymal Cells in Physiological and Pathological Conditions
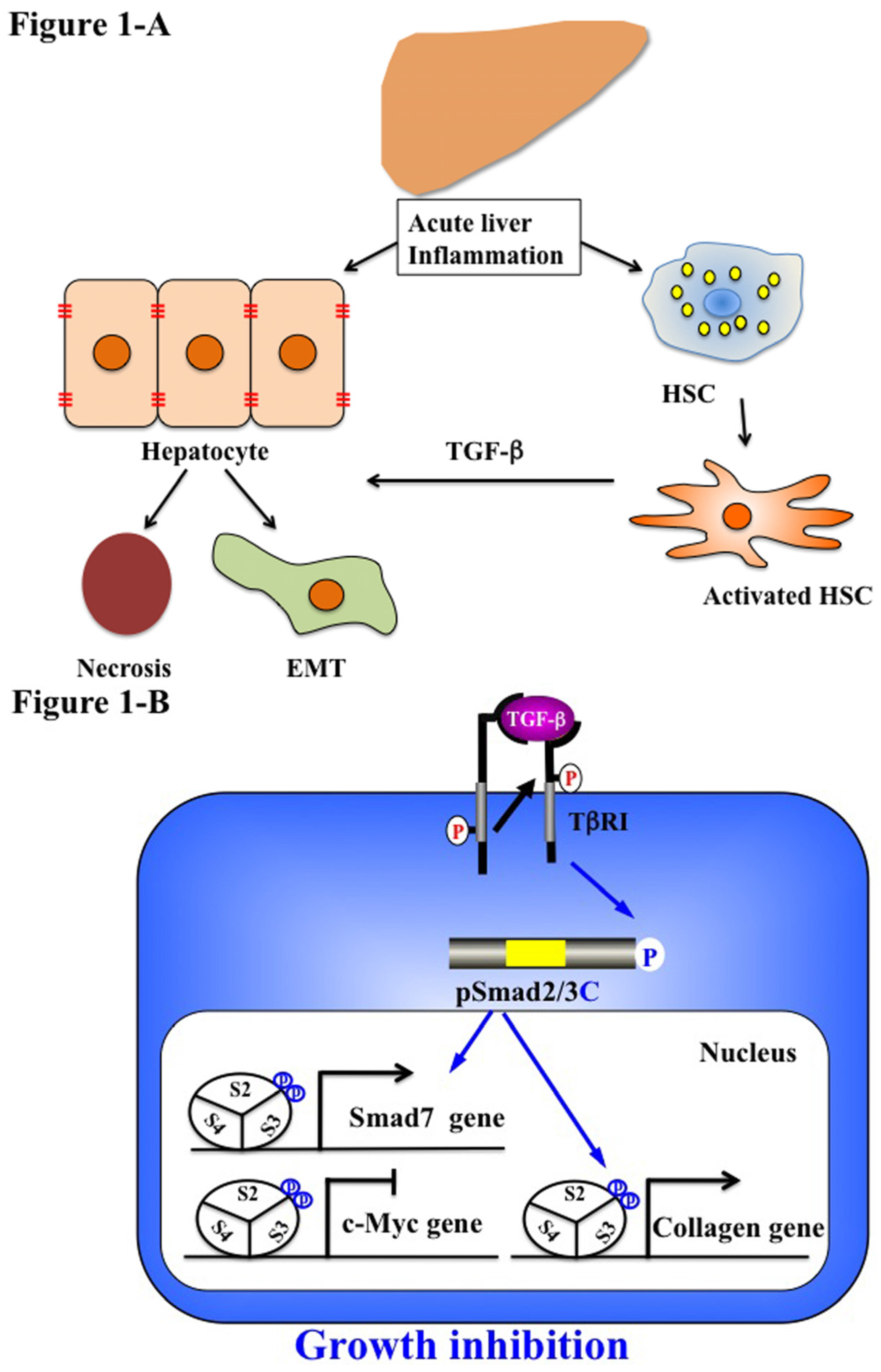
5.1. Acute Liver Injury (Physiological Type 2 EMT)

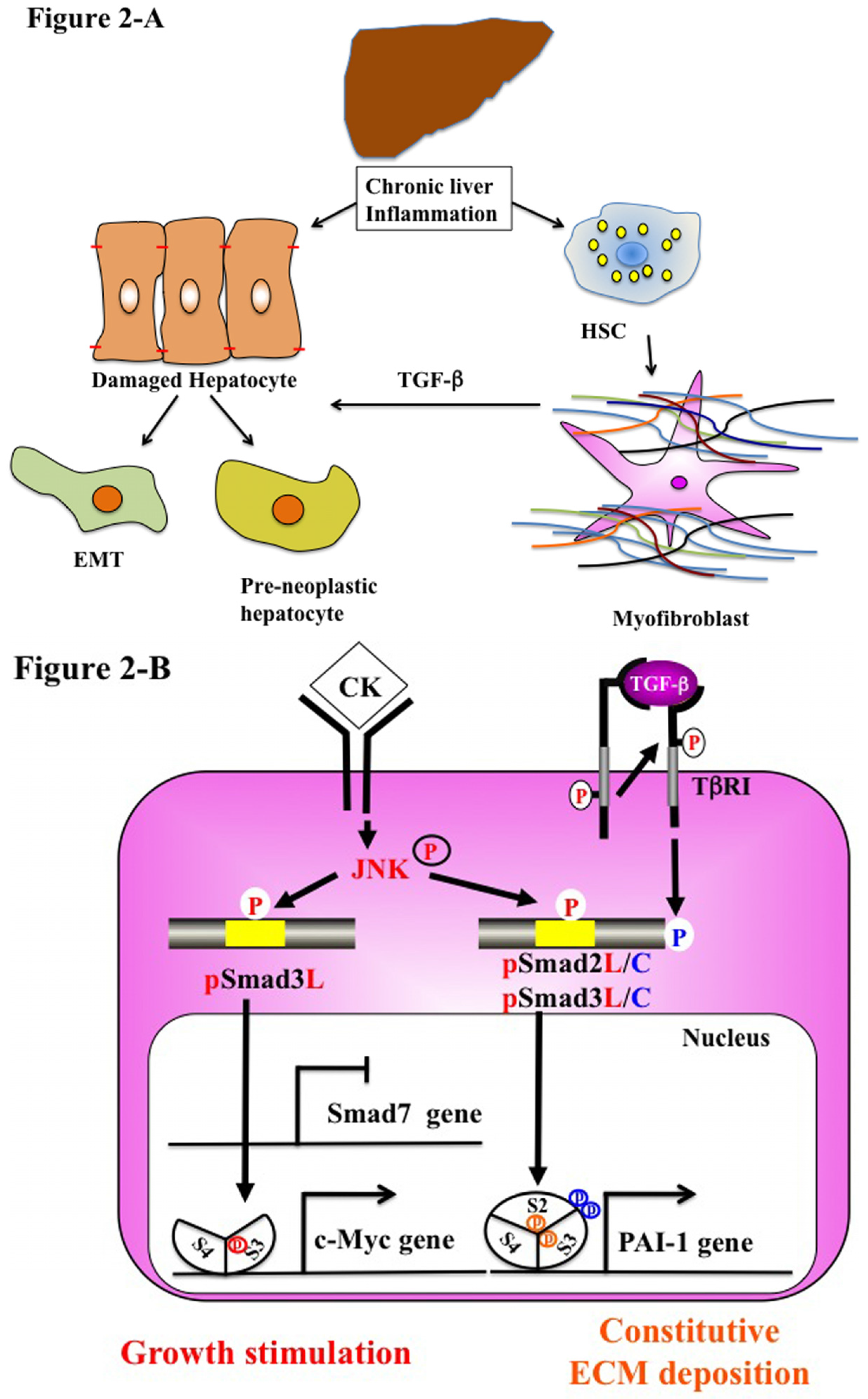
5.2. Chronic Liver Injury (Pathophysiological Type 2 EMT)

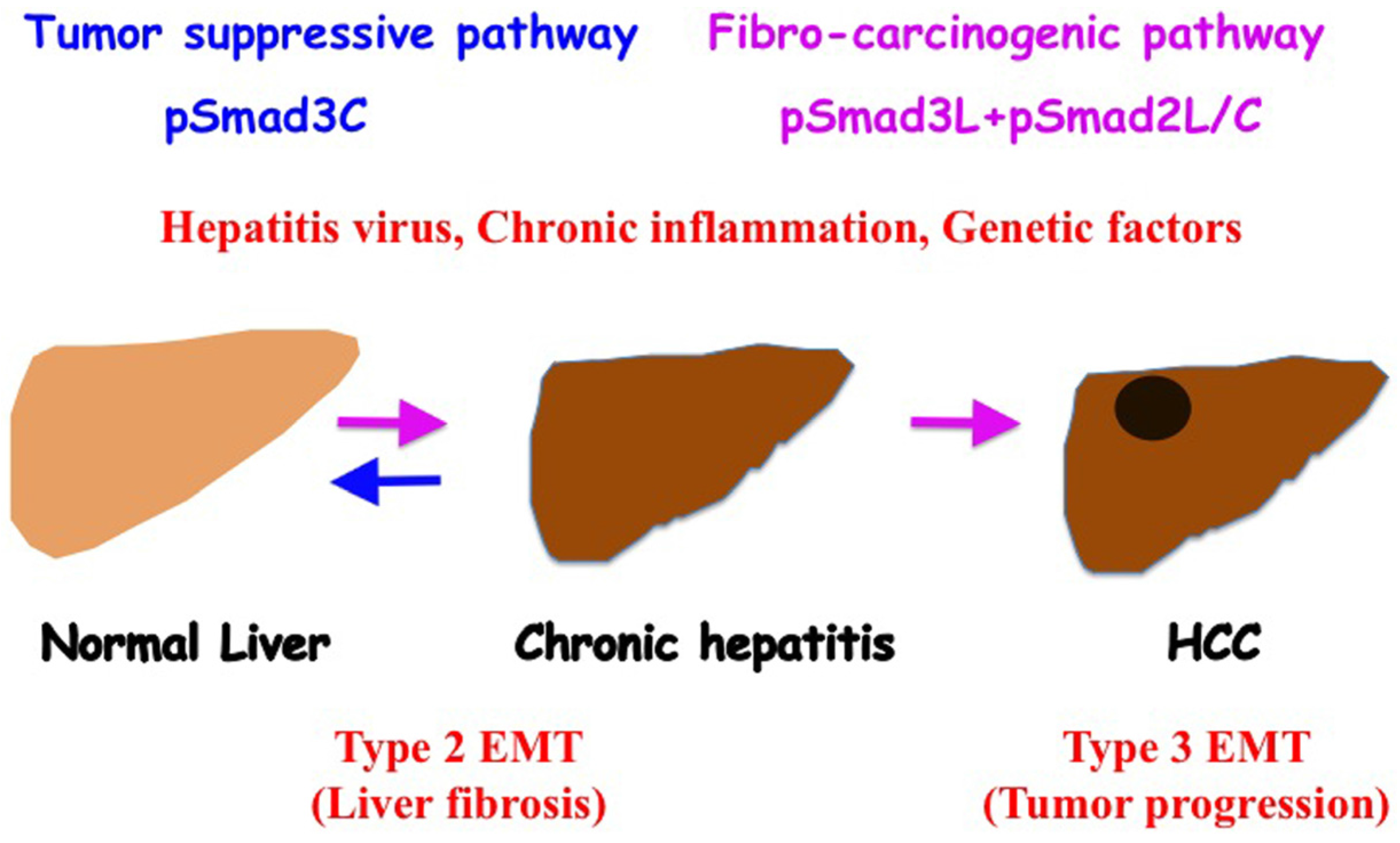
5.3. Stem Cells and Advanced HCC Biology (Type 3 EMT)
6. Reversible Human TGF-β Signal Shifting between Tumor-Suppression and Fibro-Carcinogenesis

7. Conclusions
Conflicts of Interest
References
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Serra, R. Regulation of differentiation by TGF-β. Curr. Opin. Genet. Dev. 1996, 6, 581–586. [Google Scholar] [CrossRef]
- Sporn, M.B.; Rovberts, A.B. The Transforming Growth Factor-βs; Springer: Berlin, Germany, 1990. [Google Scholar]
- Bellam, N.; Pasche, B. TGF-β signaling alterations and colon cancer. Cancer Treat Res. 2010, 155, 85–103. [Google Scholar] [PubMed]
- Matsuzaki, K. Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013, 24, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad phosphoisoform signals in acute and chronic liver injury: Similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 2012, 347, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Date, M.; Matsuzaki, K.; Matsushita, M.; Sakitani, K.; Shibano, K.; Okajima, A.; Yamamoto, C.; Ogata, N.; Okumura, T.; Seki, T.; et al. Differential expression of transforming growth factor-β and its receptors in hepatocytes and nonparenchymal cells of rat liver after CCl4 administration. J. Hepatol. 1998, 28, 572–581. [Google Scholar] [CrossRef]
- Date, M.; Matsuzaki, K.; Matsushita, M.; Tahashi, Y.; Furukawa, F.; Inoue, K. Modulation of transforming growth factor beta function in hepatocytes and hepatic stellate cells in rat liver injury. Gut 2000, 46, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N. Liver regeneration. J. Hepatol. 2000, 32, 19–31. [Google Scholar] [CrossRef]
- Palmes, D.; Spiegel, H.U. Animal models of liver regeneration. Biomaterials 2004, 25, 1601–1611. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Kojiro, M.; Roskams, T. Early hepatocellular carcinoma and dysplastic nodules. Semin. Liver Dis. 2005, 25, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Joo, K.M.; Lee, J.; Yoon, Y.; Nam, D.H. Targeting the epithelial to mesenchymal transition in glioblastoma: The emerging role of MET signaling. OncoTargets Ther. 2014, 7, 1933–1944. [Google Scholar]
- Pinzani, M. Epithelial-mesenchymal transition in chronic liver disease: Fibrogenesis or escape from death? J. Hepatol. 2011, 55, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Baum, B.; Settleman, J.; Quinlan, M.P. Transitions between epithelial and mesenchymal states in development and disease. Semin. Cell Dev. Biol. 2008, 19, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, C.; Amicone, L.; Alonzi, T.; Marchetti, A.; Mancone, C.; Tripodi, M. Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte. Liver Int. 2015, 35, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, C.; Filippini, D.; Coen, S.; Marchetti, A.; Cavallari, C.; Laudadio, I.; Spagnoli, F.M.; Alonzi, T.; Tripodi, M. Snail controls differentiation of hepatocytes by repressing HNF4α expression. J. Cell. Physiol. 2006, 209, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, A.; Potter, J.; Kaimori, J.Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-β1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.L.; Mainez, J.; Vega, S.; Sancho, P.; Murillo, M.M.; de Frutos, C.A.; Del Castillo, G.; Lopez-Blau, C.; Fabregat, I.; Nieto, M.A. Snail1 suppresses TGF-β-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J. Cell Sci. 2010, 123, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Eischeid, A.N.; Chen, X.M. Col1A1 production and apoptotic resistance in TGF-β1-induced epithelial-to-mesenchymal transition-like phenotype of 603B cells. PLoS ONE 2012, 7, e51371. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Danoff, T.M.; Kalluri, R.; Neilson, E.G. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997, 273, F563–F574. [Google Scholar] [PubMed]
- Nitta, T.; Kim, J.S.; Mohuczy, D.; Behrns, K.E. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology 2008, 48, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.; Rubino, B.; Ballabio, G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology 2007, 39, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Massague, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Hader, C.; Marlier, A.; Cantley, L. Mesenchymal-epithelial transition in epithelial response to injury: The role of Foxc2. Oncogene 2010, 29, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Gotzmann, J.; Huber, H.; Thallinger, C.; Wolschek, M.; Jansen, B.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-β1 and Ha-Ras: Steps towards invasiveness. J. Cell Sci. 2002, 115, 1189–1202. [Google Scholar] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Miura, K.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Penz-Osterreicher, M.; Brenner, D.A. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 2010, 51, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.; Osterreicher, C.H.; Scholten, A.; Iwasako, K.; Gu, G.; Brenner, D.A.; Kisseleva, T. Genetic labeling dose not detect epithelial-to-mesechymal transition of cholangiocytes in liver fibrosis in mice. Gatroenterology 2010, 139, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Diehl, A.M. Evidence for and against epithelial-to mesenchymal transition in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G881–G890. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Kruger, L.; Premont, R.; Yang, L.; Syn, W.K.; Metzger, D.; et al. Smoothened is a master regulator of adult liver repair. J. Clin. Investig. 2013, 23, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Kisseleva, T.; Iwaisako, K.; Miura, K.; Taura, K.; de Minicis, S.; Osterreicher, C.H.; Schnabl, B.; Seki, E.; Brenner, D.A. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 2009, 137, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L. Regulation of Smad activity. Cell 2000, 100, 189–192. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFβ/ Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, G.; Matsuzaki, K.; Yoshida, K.; Mori, S.; Murata, M.; Seki, T.; Matsui, H.; Fujisawa, J.; Okazaki, K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007, 67, 5090–5096. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Kitano, C.; Murata, M.; Sekimoto, G.; Yoshida, K.; Uemura, Y.; Seki, T.; Taketani, S.; Fujisawa, J.; Okazaki, K. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-β signal in later stages of human colorectal cancer. Cancer Res. 2009, 69, 5321–5330. [Google Scholar] [CrossRef] [PubMed]
- Kamaraju, A.K.; Roberts, A.B. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 2005, 280, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Matsuzaki, K.; Yoshida, K.; Furukawa, F.; Tahashi, Y.; Yamagata, H.; Sekimoto, G.; Seki, T.; Matsui, H.; Nishizawa, M.; et al. TGFβ and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 2004, 23, 7416–7429. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad3 phosphoisoform-mediated signaling during sporadic human colorectal carcinogenesis. Histol. Histopathol. 2006, 21, 645–662. [Google Scholar] [PubMed]
- Sapkota, G.; Knockaert, M.; Alarcon, C.; Montalvo, E.; Brivanlou, A.H.; Massague, J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 2006, 281, 40412–40419. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGFβ superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, H.; Matsuzaki, K.; Mori, S.; Yoshida, K.; Tahashi, Y.; Furukawa, F.; Sekimoto, G.; Watanabe, T.; Uemura, Y.; Sakaida, N.; et al. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH2-terminal kinase during human colorectal carcinogenesis. Cancer Res. 2005, 65, 157–165. [Google Scholar] [PubMed]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGFβ pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Matsuzaki, K.; Yoshida, K.; Sekimoto, G.; Tahashi, Y.; Mori, S.; Uemura, Y.; Sakaida, N.; Fujisawa, J.; Seki, T.; et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-β signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 2009, 49, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGFβ signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGFβ signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-Smad TGFβ signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, M. The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGFβ receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGFβ. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad phosphoisoform signaling specificity: The right place at the right time. Carcinogenesis 2011, 32, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massague, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGFβ signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yamagata, H.; Furukawa, F.; Seki, T.; Nishizawa, M.; Fujisawa, J.; Okazaki, K. Transforming growth factor-β and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 2005, 166, 1029–1039. [Google Scholar] [CrossRef]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled receptor kinase-2 is a TGFβ-inducible antagonist of TGFβ signal transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.J.; Piek, E.; Heyer, J.; Escalante-Alcalde, D.; Stewart, C.L.; Weinstein, M.; Deng, C.; Kucherlapati, R.; Bottinger, E.P.; Roberts, A.B. Functional characterization of transforming growth factor β signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem. 2001, 276, 19945–19953. [Google Scholar]
- Feng, X.; Zhang, Y.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGFβ response. Nature 1996, 383, 168–172. [Google Scholar]
- Sun, Y.; Liu, X.; Constantinescu, S.N.; Karam, E.; Weinberg, R.A.; Lodish, H.F. Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10669–10674. [Google Scholar]
- Alcorn, J.F.; Velden, J.L.; Guala, A.S.; Badura, E.C.; Janssen-Heininger, Y.M. c-Jun N-terminal kinase 1 promotes transforming growth factor-β1-induced epithelial-to-mesenchymal transition via control of linker phosphorylation and transcriptional activity of Smad3. Am. J. Respir. Cell Mol. Biol. 2011, 44, 571–581. [Google Scholar]
- Kobayashi, H.; Hirashima, Y.; Suzuki, M.; Tanaka, Y.; Kanayama, N.; Terao, T. Transforming growth factor-β1 produced by ovarian cancer cell line HRA stimulates attachment and invasion through an up-regulation of plasminogen activator inhibitor type-1 in human peritoneal mesothelial cells. J. Biol. Chem. 2003, 278, 26793–26802. [Google Scholar]
- Hu, P.F.; Chen, H.; Zhong, W.; Lin, Y.; Zhang, X.; Chen, Y.X.; Xie, W.F. Adenovirus-mediated transfer of siRNA against PAI-1 mRNA ameliorates hepatic fibrosis in rats. J. Hepatol. 2009, 51, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Miyazono, K. Roles of TGF-β family signaling in stem cell renewal and differentiation. Cell Res. 2009, 19, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Ajima, R.; Luo, C.T.; Yamaguchi, T.P.; Stappenbeck, T.S. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science 2012, 338, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Kishimoto, M.; Nakajima, A.; Yamashina, M.; Nakayama, S.; Kusuda, T.; Sakaguchi, Y.; Yoshida, K.; Uchida, K.; Nishio, A.; et al. The specific linker phosphorylation of Smad2/3 indicates epithelial stem cells in stomach; particularly increasing in mucosae of Helicobacter-associated gastritis. J. Gastroenterol. 2011, 46, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, M.; Fukui, T.; Suzuki, R.; Takahashi, Y.; Sumimoto, K.; Okazaki, T.; Sakao, M.; Sakaguchi, Y.; Yoshida, K.; Uchida, K.; et al. Phosphorylation of Smad2/3 at specific linker threonine indicates slow-cycling intestinal stem-like cells before reentry to cell cycle. Dig. Dis. Sci. 2015, 60, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.R.; Yang, C.Q.; Shi, Q. Transforming growth factor-β1 induced epithelial–mesenchymal transition in liver fibrosis. Hepatogastroenterology 2012, 59, 1960–1963. [Google Scholar] [PubMed]
- Dooley, J.S.; Lok, A.; Burroughs, A.K.; Heathcote, J. Sherlock’s Disease of the Liver and Biliary System; Wiley-Blackwell: West Sussex, UK, 2011. [Google Scholar]
- Tahashi, Y.; Matsuzaki, K.; Date, M.; Yoshida, K.; Furukawa, F.; Sugano, Y.; Matsushita, M.; Himeno, Y.; Inagaki, Y.; Inoue, K. Differential regulation of TGF-β signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 2002, 35, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K. Differential Regulation of TGF-β/Smad Signaling in Hepatic Stellate Cells between Acute and Chronic Liver Injuries. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat. Clin. Pract. Gastroenterol. Hepatol. 2004, 1, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Housset, C.N.; Friedman, S.L. Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. J. Clin. Investig. 1993, 92, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.A.; Waterboer, T.; Choi, S.K.; Lindquist, J.N.; Stefanovic, B.; Burchardt, E.; Yamauchi, M.; Gillan, A.; Rippe, R.A. New aspects of hepatic fibrosis. J. Hepatol. 2000, 32, 32–38. [Google Scholar] [CrossRef]
- Marra, F. Chemokines in liver inflammation and fibrosis. Front. Biosci. 2002, 7, 899–914. [Google Scholar] [CrossRef]
- Reimann, T.; Hempel, U.; Krautwald, S.; Axmann, A.; Scheibe, R.; Seidel, D.; Wenzel, K.W. Transforming growth factor-β1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett. 1997, 403, 57–60. [Google Scholar] [CrossRef]
- Pinzani, M.; Gesualdo, L.; Sabbah, G.M.; Abboud, H.E. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J. Clin. Investig. 1989, 84, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Fouassier, L.; Chung, J.J.; Carayon, A.; Vallee, P.; Rey, C.; Housset, C. Cellular localization of endothelin-1 and increased production in liver injury in the rat: Potential for autocrine and paracrine effects on stellate cells. Hepatology 1998, 27, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Arrighi, M.C.; Fazi, M.; Caligiuri, A.; Pinzani, M.; Romanelli, R.G.; Efsen, E.; Laffi, G.; Gentilini, P. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor’s actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology 1999, 30, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Nouchi, T.; Tanaka, Y.; Tsukada, T.; Sato, C.; Marumo, F. Appearance of α-smooth-muscle-actin-positive cells in hepatic fibrosis. Liver 1991, 11, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Graff, A.; Kruger, S.; Bochard, F.; Gabbiani, G.; Denk, H. Modulation of α smooth muscle actin and desmin expression in perisinusoidal cells of normal and diseased human livers. Am. J. Pathol. 1991, 138, 1233–1242. [Google Scholar] [PubMed]
- Dooley, S.; Hamzavi, J.; Breitkopf, K.; Wiercinska, E.; Said, H.M.; Lorenzen, J.; Ten Dijke, P.; Gressner, A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003, 125, 178–191. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, J.; Ma, R.; Lin, H.; Liang, X.; Cai, X. Prognostic significance of E-cadherin expression in hepatocellular carcinoma: A meta-analysis. PLoS ONE 2014, 9, e103952. [Google Scholar] [CrossRef] [PubMed]
- Muta, H.; Noguchi, M.; Kanai, Y.; Ochiai, A.; Nawata, H.; Hirohashi, S. E-cadherin gene mutations in signet ring cell carcinoma of the stomach. Jpn. J. Cancer Res.: Gann 1996, 87, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Kanai, Y.; Maesawa, C.; Ochiai, A.; Torii, A.; Hirohashi, S. Disruption of E-cadherin-mediated cell adhesion systems in gastric cancers in young patients. Jpn. J. Cancer Res.: Gann 1999, 90, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Zanudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P., Jr.; Albert, R. Network modeling of TGFβ signaling in hepatocellular carcinoma epithelial-to-mesenchymal transition reveals joint sonic hedgehog and Wnt pathway activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Heider, K.H.; Beug, H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr. Biol.: CB 1998, 8, 1243–1252. [Google Scholar] [CrossRef]
- Weng, H.L.; Liu, Y.; Chen, J.L.; Huang, T.; Xu, L.J.; Godoy, P.; Hu, J.H.; Zhou, C.; Stickel, F.; Marx, A.; et al. The etiology of liver damage imparts cytokines transforming growth factor β1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 2009, 50, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Theise, N.D.; Saxena, R.; Portmann, B.C.; Thung, S.N.; Yee, H.; Chiriboga, L.; Kumar, A.; Crawford, J.M. The canals of hering stem cells in humans. Hepatology 1999, 30, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.C.; Evarts, R.P.; Nakatsukasa, H.; Marsden, E.R.; Thorgeirsson, S.S. Occurrence of oval-type cells in hepatitis B virus-associated human hepatocarcinogenesis. Hepatology 1992, 16, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor β gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-β1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, A.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.; Fernandez-Rodriguez, C.M.; et al. A mesenchymal-like phenotype and expression of CD44 predict lack of apoptotic response to sorafenib in liver tumor cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W. Reversibility of oncogene-induced cancer. Curr. Opin. Genet. Dev. 2004, 14, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Matsuzaki, K.; Inokuchi, R.; Kawamura, R.; Yoshida, K.; Murata, M.; Fujisawa, J.; Fukushima, N.; Sata, M.; Kage, M.; et al. Phosphorylated Smad2 and Smad3 signaling: Shifting between tumor suppression and fibro-carcinogenesis in chronic hepatitis C. Hepatol. Res. 2013, 43, 1327–1342. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.R.; Yoshida, K.; Jin, Q.; Murata, M.; Yamaguchi, T.; Tsuneyama, K.; Moritoki, Y.; Niu, J.; Matsuzaki, K.; Lian, Z.X. Reversible phospho-Smad3 signaling between tumor-suppression and fibro-carcinogenesis in chronic hepatitis B infection. Clin. Exp. Immunol. 2014, 176, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, S.; Llovet, J.M. New molecular therapies for hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S80–S85. [Google Scholar] [CrossRef] [PubMed]
- Nakakura, E.K.; Choti, M.A. Management of hepatocellular carcinoma. Oncology (Williston Park) 2000, 14, 1085–1098, discussion 1098–1102. [Google Scholar] [PubMed]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 2014, 20, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, R.; Maynard, E.; Shah, M.; Daily, M.F.; Chen, C.; Spear, B.T.; Gedaly, R. Synergistic inhibition of HCC and liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and WNT/β-catenin pathways. Anticancer Res. 2014, 34, 1709–1713. [Google Scholar] [PubMed]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Akhurst, R.J.; Balmain, A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat. Cell Biol. 2002, 4, 487–494. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, K.; Murata, M.; Yamaguchi, T.; Matsuzaki, K.; Okazaki, K. Reversible Human TGF-? Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. J. Clin. Med. 2016, 5, 7. https://doi.org/10.3390/jcm5010007
Yoshida K, Murata M, Yamaguchi T, Matsuzaki K, Okazaki K. Reversible Human TGF-? Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. Journal of Clinical Medicine. 2016; 5(1):7. https://doi.org/10.3390/jcm5010007
Chicago/Turabian StyleYoshida, Katsunori, Miki Murata, Takashi Yamaguchi, Koichi Matsuzaki, and Kazuichi Okazaki. 2016. "Reversible Human TGF-? Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions" Journal of Clinical Medicine 5, no. 1: 7. https://doi.org/10.3390/jcm5010007
APA StyleYoshida, K., Murata, M., Yamaguchi, T., Matsuzaki, K., & Okazaki, K. (2016). Reversible Human TGF-? Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. Journal of Clinical Medicine, 5(1), 7. https://doi.org/10.3390/jcm5010007