
Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cell Culture
2.3. Cell Proliferation Assays
2.4. Cell Migration Assays
2.5. Cell Incubations for Signal Transduction Assays
2.6. Reverse Transcription Polymerase Chain Reacton (RT-PCR)
2.7. Immunoblotting
2.8. Statistical Analysis
3. Results and Discussion
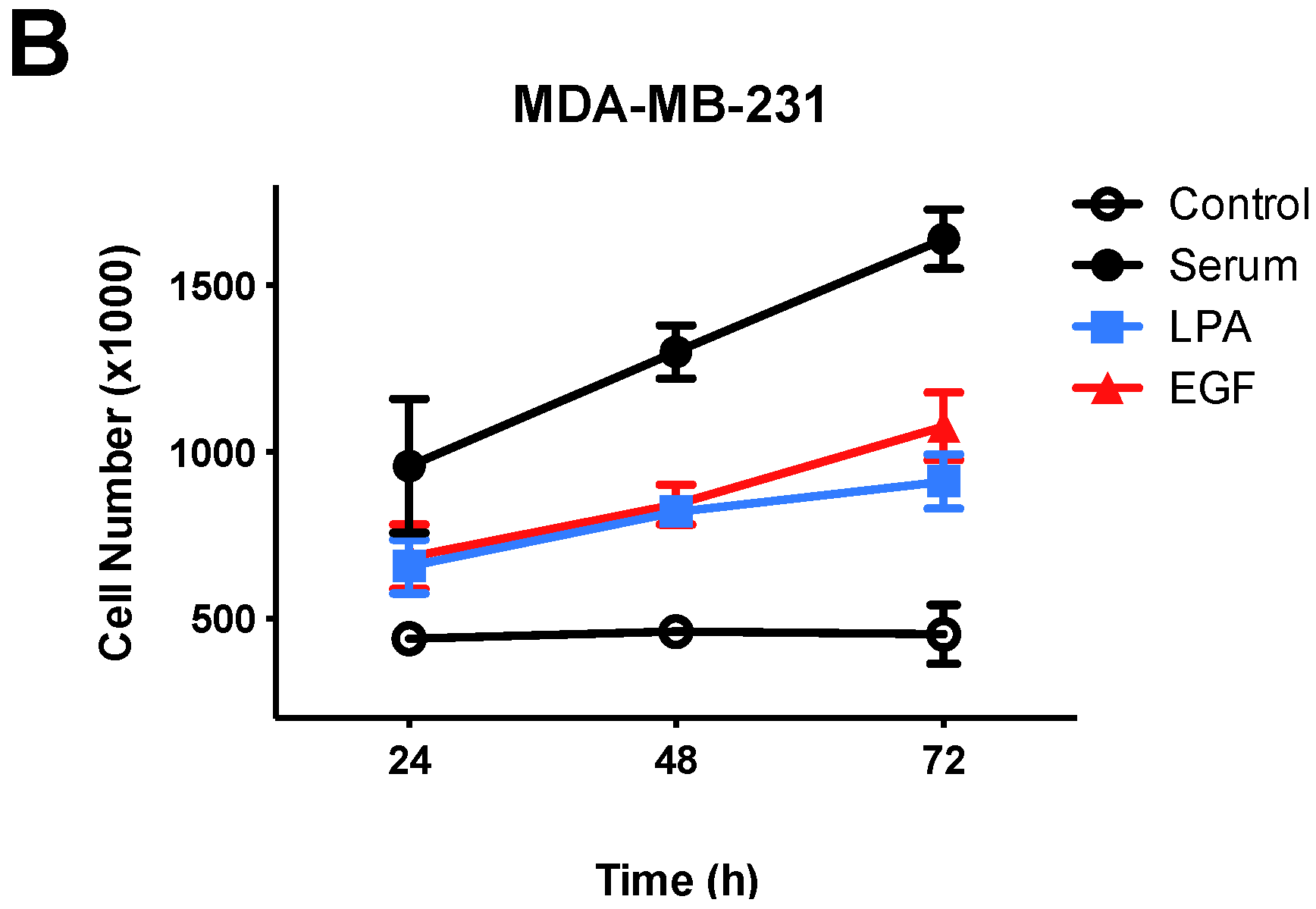
3.1. Effects of Lysophosphatidic Acid (LPA) and Epidermal Growth Factor (EGF) on Breast Cancer Cell Proliferation


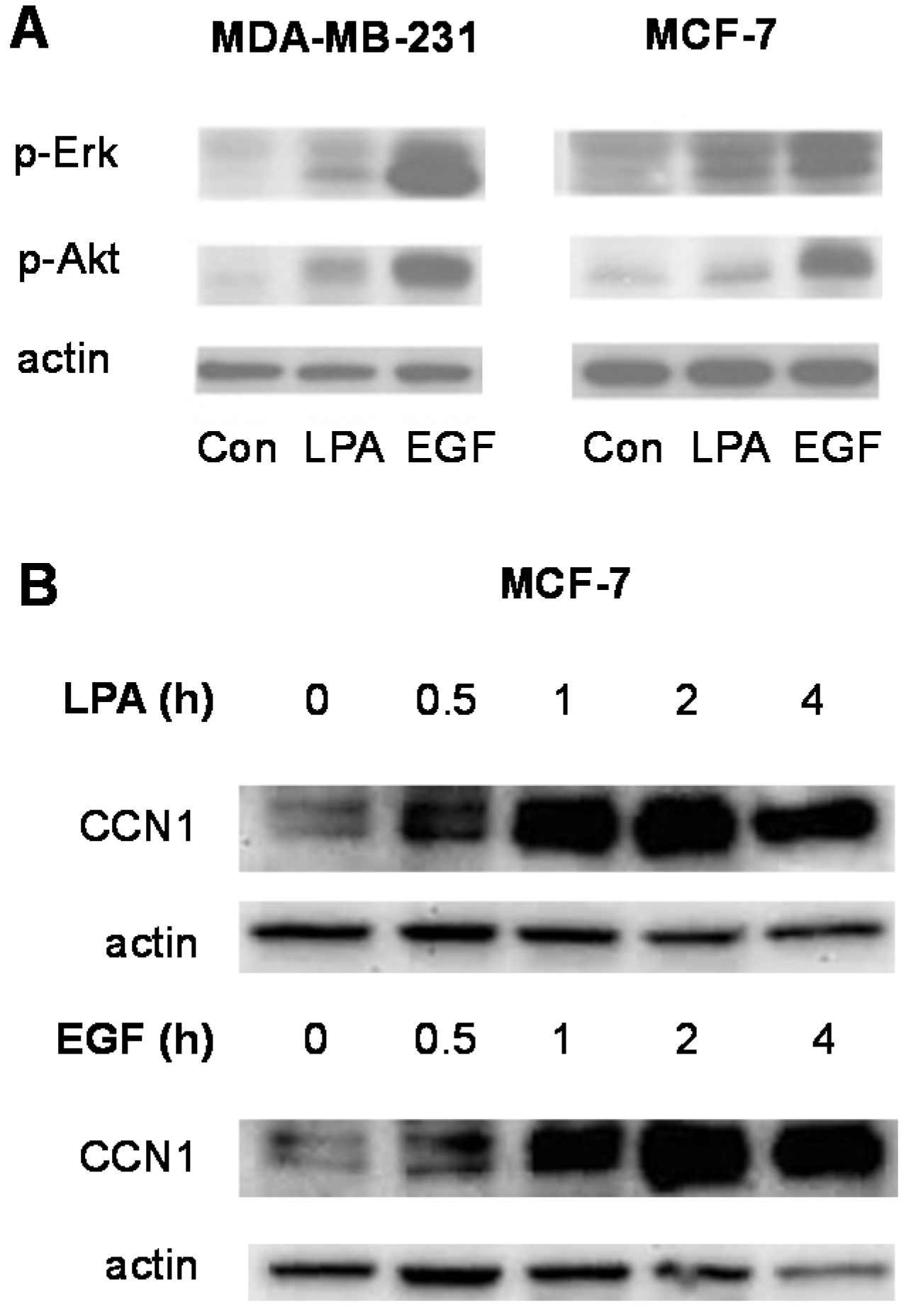
3.2. Signal Transduction Responses to LPA and EGF in Breast Cancer Cells

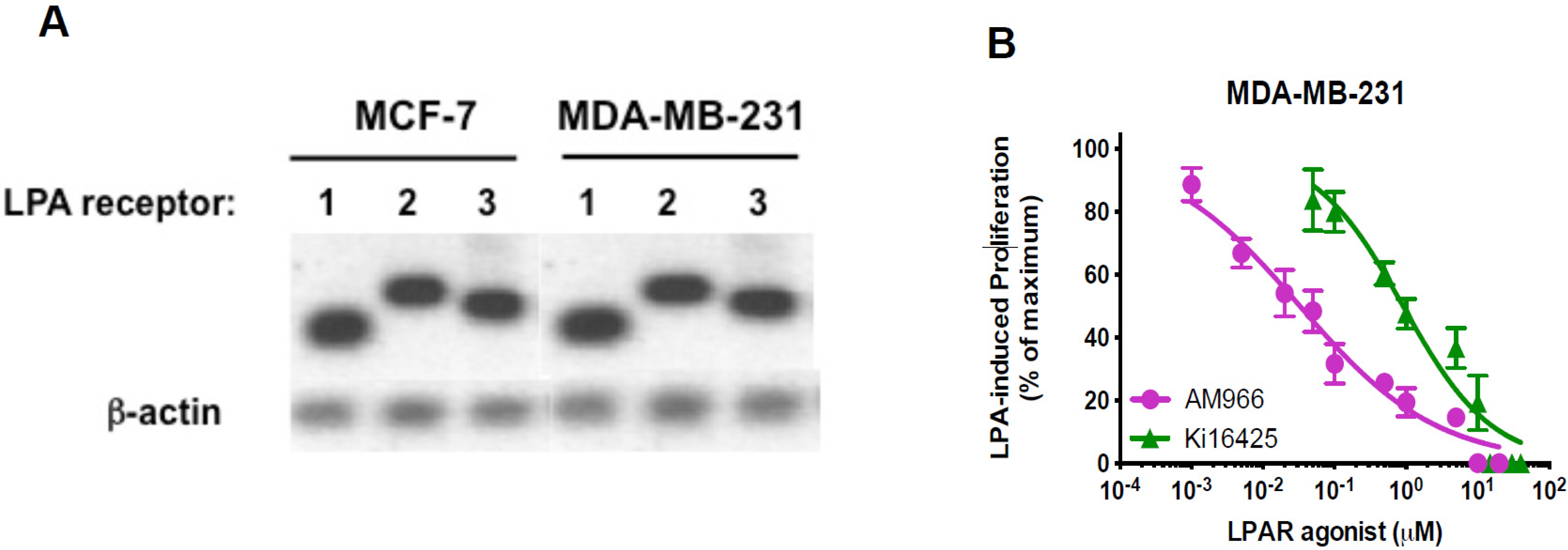
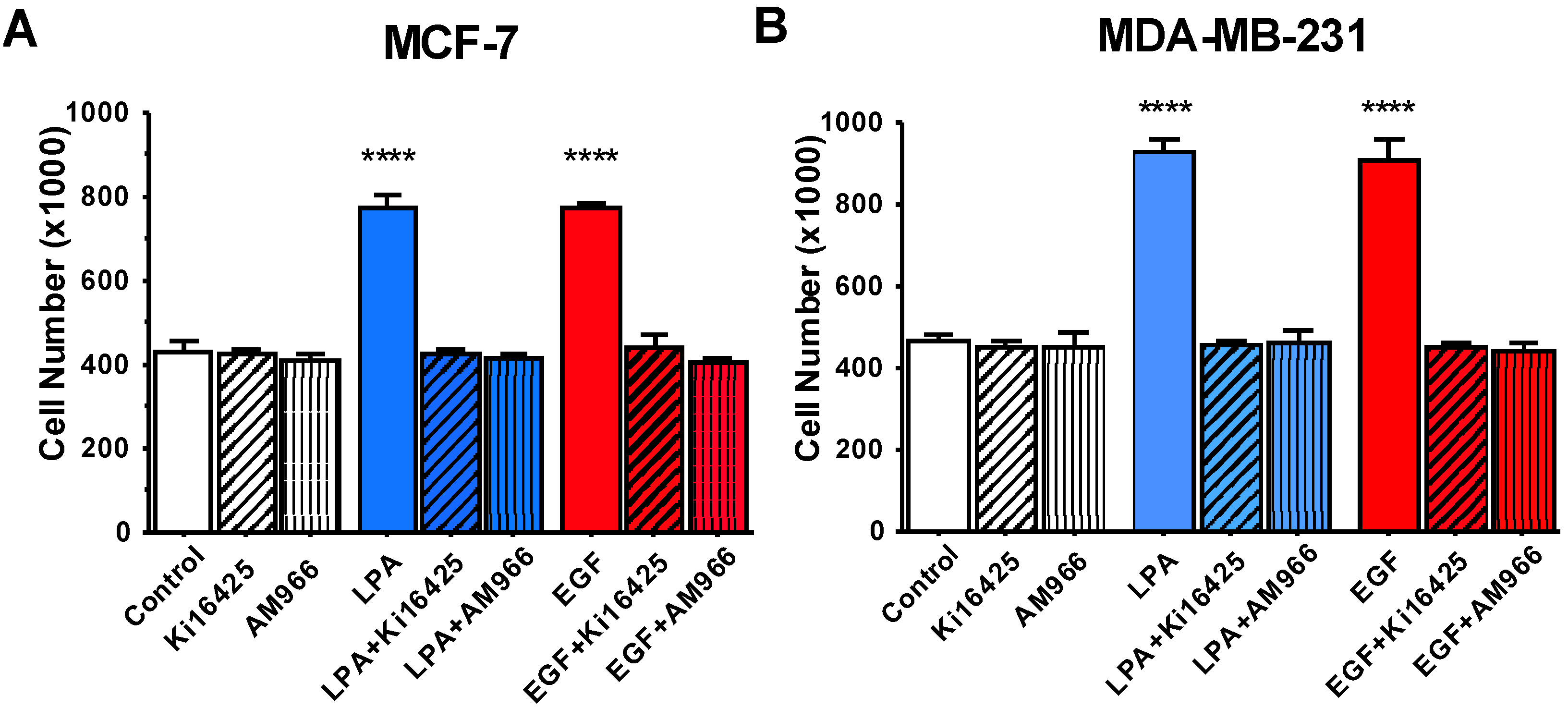
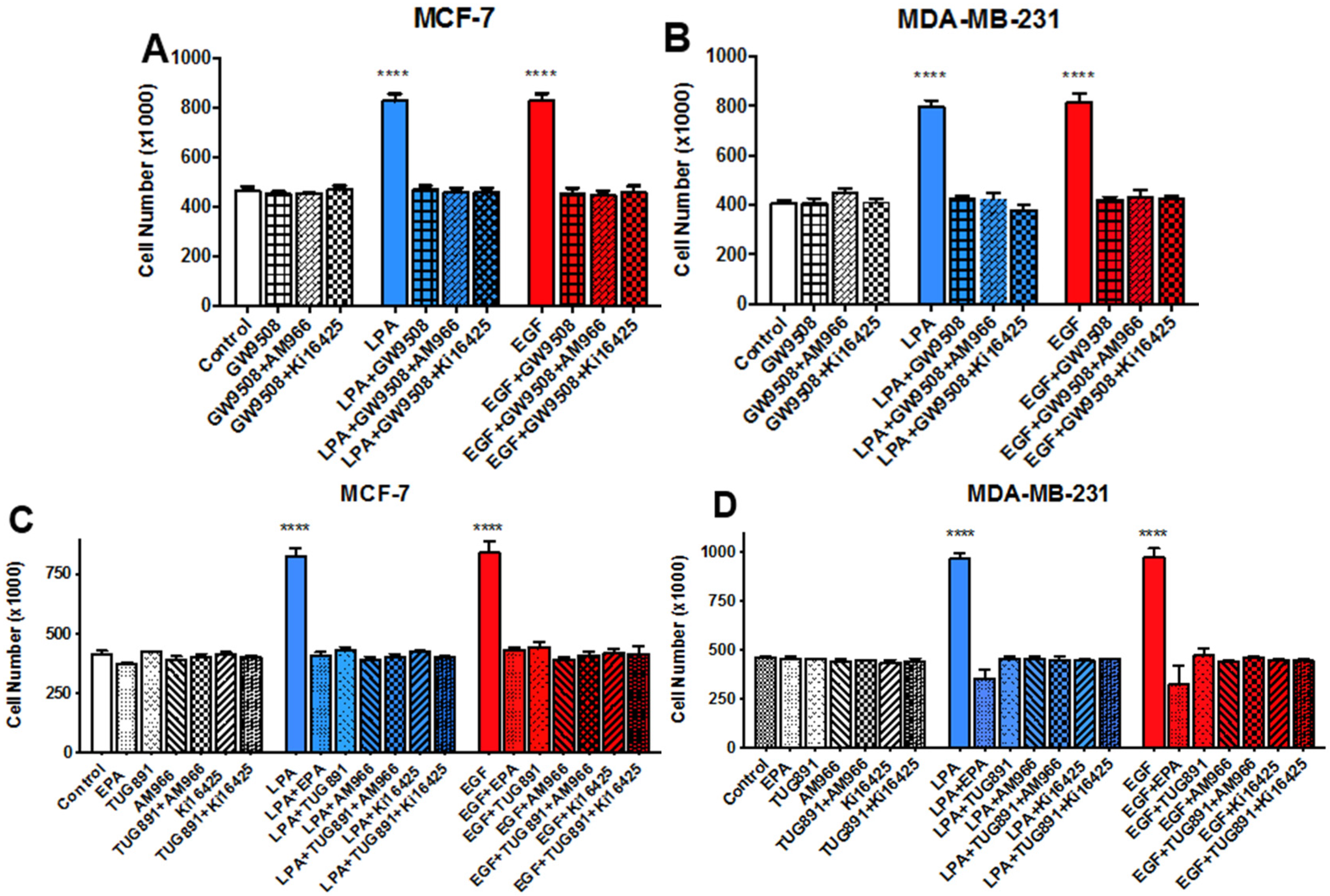
3.3. Effects of LPA Antagonists on Breast Cancer Cell Proliferation


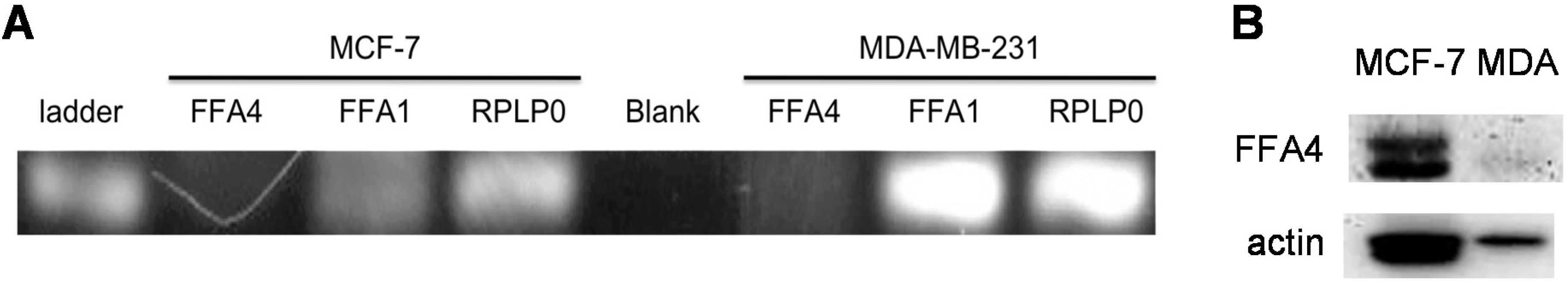
3.4. Expression of Free Fatty Acid Receptors (FFARs) in Breast Cancer Cells

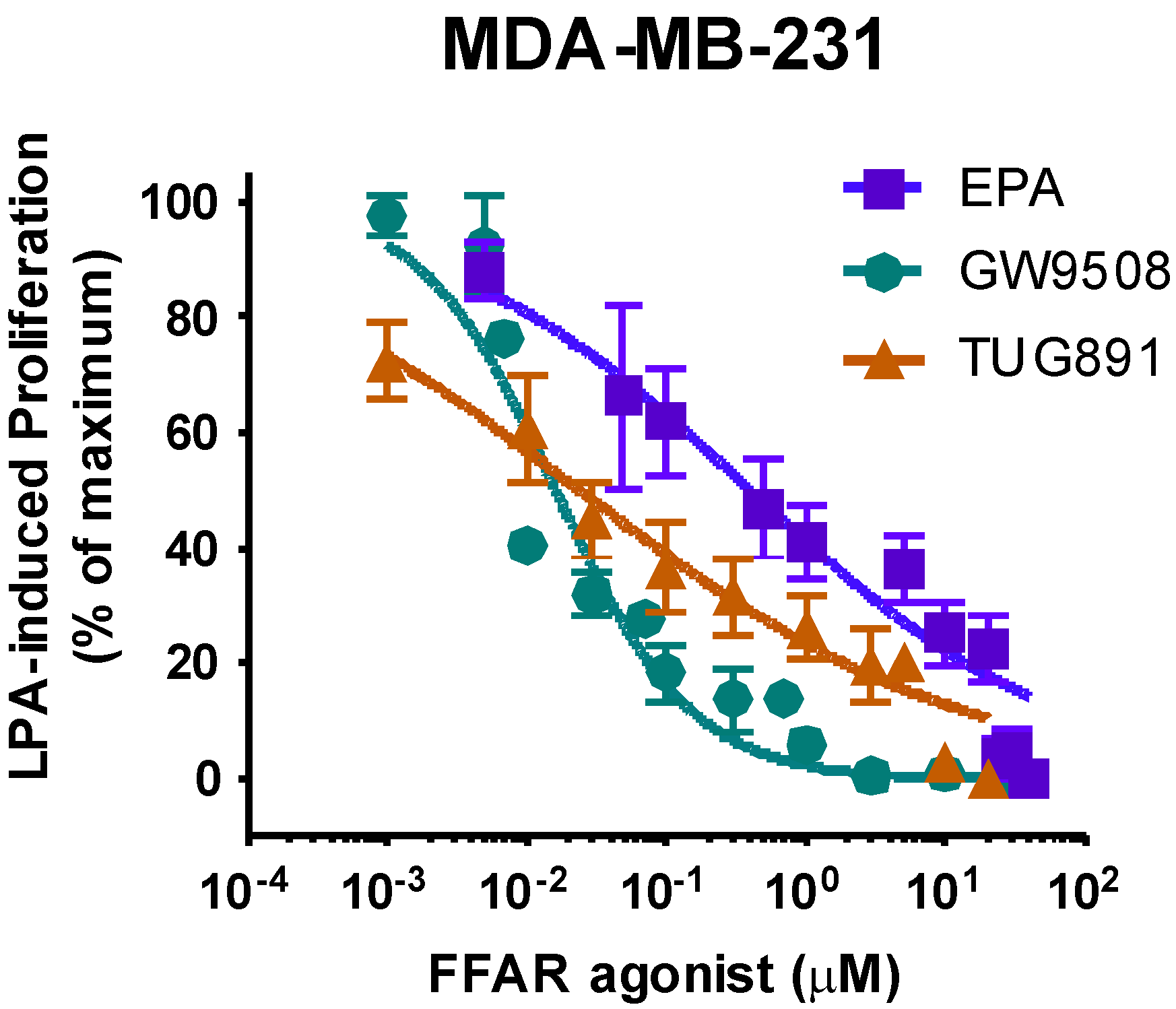
3.5. Effects of FFAR Agonists on Breast Cancer Cell Proliferation


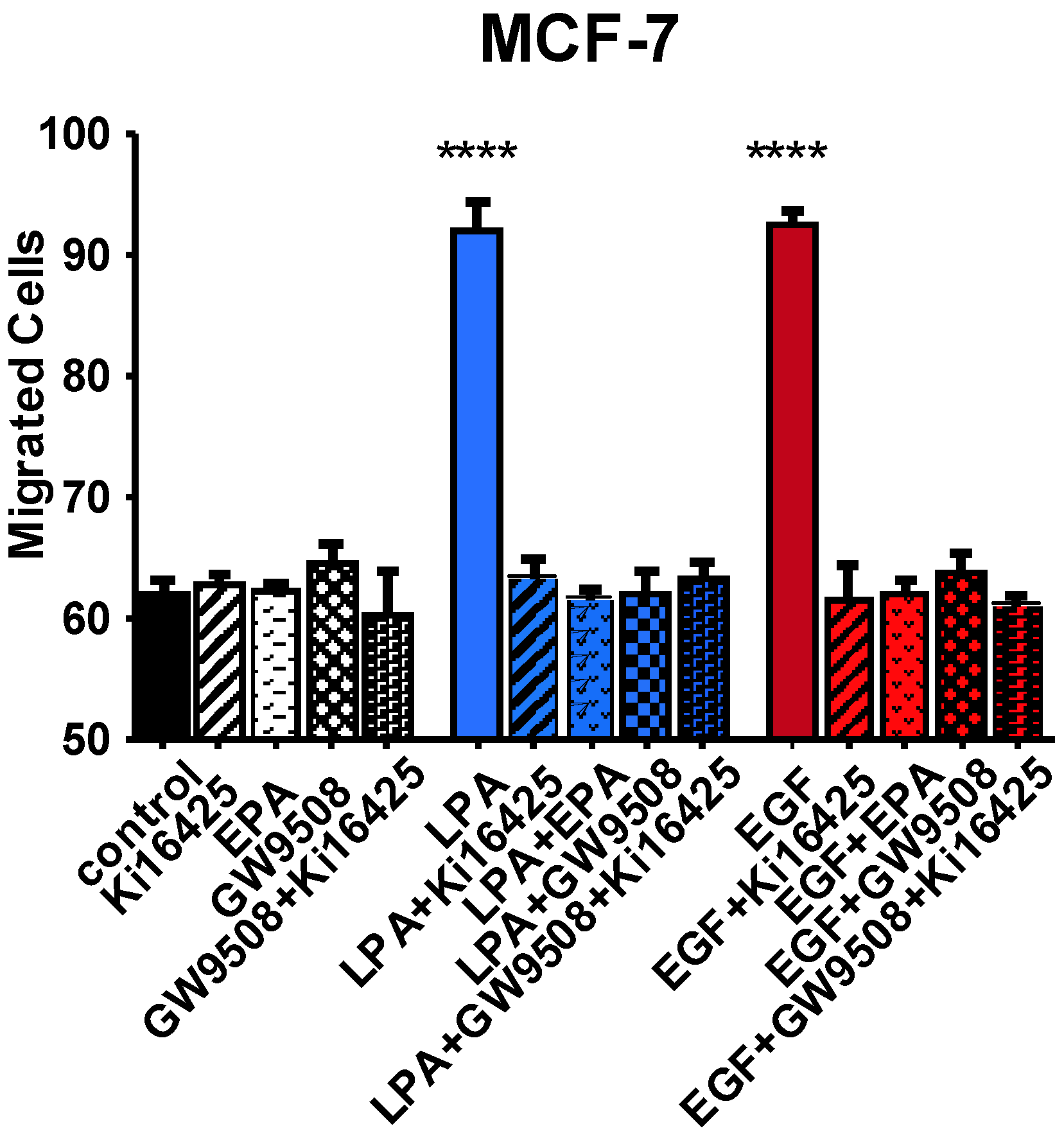
3.6. Effects of FFAR Agonists on Breast Cancer Cell Migration

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, Z.; Hopkins, M.M.; Zhang, Z.; Quisenberry, C.B.; Fix, L.C.; Galvan, B.M.; Meier, K.E. ω-3 Fatty acids and other FFA4 agonists inhibit growth factor signaling in human prostate cancer cells. J. Pharm. Exp. Ther. 2015, 352, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Meier, K.E. ω-3 Fatty acids and their impact on prostate cancer risk. Curr. Nutr. Rep. 2015, in press. [Google Scholar]
- Hardman, W.E. ω-3 Fatty acids to augment cancer therapy. J. Nutr. 2002, 132, 3509S–3512S. [Google Scholar]
- Zheng, J.S.; Hu, X.J.; Zao, Y.M.; Yang, J.; Li, D. Intake of fish and marine n-3 polyunsaturated fatty acids and risk of breast cancer: Meta-analysis of data from 21 Independent Prospective Cohort Studies. Br. Med. J. 2013, 346, f3706. [Google Scholar] [CrossRef] [PubMed]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of ω-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Pogash, T.J.; el-Bayoumy, K.; Amin, S.; Gowda, K.; de Cicco, R.L.; Barton, M.; Su, Y.; Russo, I.H.; Himmelberger, J.A.; Slifker, M.; et al. Oxidized derivative of docosahexaenoic acid preferentially inhibit cell proliferation in triple negative over luminal breas cancer cells. Vitro Cell Dev. Biol. Anim. 2015, 52, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Karmali, R.A.; Marsh, J.; Ruchs, C. Effect of ω-3 fatty acids on growth of a rat mammary tumor. J. Natl. Cancer Inst. 1984, 73, 457–461. [Google Scholar] [PubMed]
- Rose, D.P.; Connolly, J.M. Effects of dietary ω-3 fatty acids on human breast cancer growth and metastases in nude mice. J. Natl. Caner Inst. 1993, 85, 1743–1747. [Google Scholar] [CrossRef]
- Sauer, L.A.; Dauchy, R.T.; Blask, D.E.; Krause, J.A.; Davidson, L.K.; Dauchy, E.M. Eicosapenaenoic acid suppresses cell proliferation in MCF-7 human breast cancer xenografts in nude rats via a pertussis toxin-sensitive signal transduction pathway. J. Nutr. 2005, 135, 2124–2129. [Google Scholar] [PubMed]
- Jiang, W.; Zhu, Z.; McGinley, J.N.; el Bayouny, K.; Manni, A.; Thompson, J.H. Identification of a molecular signature underlying inhibition of mammary carcinoma growth by dietary n-3 fatty acids. Cancer Res. 2012, 72, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Patel, S.; Jain, K. The what, where, and how of resolvins. AsPac J. Mol. Biol. Biotechnol. 2010, 20, 45–54. [Google Scholar]
- Zhang, M.J.; Spite, M. Resolvins: Anti-inflammatory and pro-resolving mediators derived from ω-3 polyunsaturated fatty acids. Annu. Rev. Nutr. 2012, 32, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Mohammed, A.; Rao, C.V. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011, 30, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaubai, N.; Johnstone, C.N.; Leong, M.M.; Li, J.; Rizzacasa, M.; Stewart, A.G. Resolvin D2 supports MCF-7 cell proliferation via activation of estrogen receptor. J. Pharmacol. Exp. Ther. 2014, 351, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.Q.; Li, P.; Wendell, J.L.; Watkins, S.M.; Olefsky, J.M. GRP120 is an ω-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2012, 143, 687–698. [Google Scholar]
- Hara, T.; Hirasawa, A.; Ichimura, A.; Kimura, I.; Tsujimoto, G. Free fatty acid receptors FFAR1 and GPR120 as novel therapeutic targets for metabolic disorders. J. Pharm. Sci. 2011, 100, 3594–3601. [Google Scholar] [CrossRef] [PubMed]
- Holliday, N.D.; Watson, S.J.; Brown, A.J.H. Drug discovery opportunities and challenges at G protein coupled receptors for long chain free fatty acids. Front. Endocrinol. 2011, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dranse, H.J.; Kelly, M.E.; Hudson, B.D. Drugs or diet? Developing novel therapeutic strategies targeting the free fatty acid family of GPCRs. Br. J. Pharmacol. 2013, 170, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.D.; Shimpukade, B.; Mackenzie, A.E.; Butcher, A.J.; Pediani, J.D.; Christiansen, E.; Heathcote, H.; Tobin, A.B.; Ulven, T.; Milligan, G. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol. Pharmacol. 2013, 84, 710–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Harvey, K.A.; Ruzmetov, N.; Welch, Z.R.; Sech, L.; Jackson, K.; Stillwell, W.; Zaloga, G.P. Siddiqui RA (2005) ω-3 polyunsaturated fatty acids attenuate breast cancer growth through activation of a neutral sphingomyelinase-mediated pathway. Int. J. Cancer 2015, 117, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gallon, D.J.; Vissac-Sabatier, C.; Antoine-Vincent, D.; Rio, P.G.; Maruizis, J.C.; Fustier, P.; Bignon, Y.J. Differential effects of n-3 and n-6 polyunsaturated fatty acids on BRCA1 and BRCA2 gene expression in breast cell lines. Br. J. Nutr. 2002, 87, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, T.; Mandall, C.C.; Woodruff, K.; St. Clair, P.; Fernandes, G.; Chouhury, G.; Ghosh-Choudhury, N. Fish oil targets PTEN to regulate NFκB for downregulation of anti-apoptotic genes in breast tumor growth. Breast Cancer Res. Treat. 2009, 118, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Monk, J.M.; Turk, H.F.; Liddle, D.M.; de Boer, A.A.; Power, K.A.; Ma, D.W.; Robinson, L.E. n-3 Polyunsaturated fatty acids and mechanisms to mitigate inflammatory paracrine signaling in obesity—Associated breast cancer. Nutrients 2014, 5, 4760–4793. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, T.; Katoh, K.; Obara, Y. Existence of GPR40 functioning in a human breast cancer cell line, MCF-7. Biochem. Biophys. Res. Commun. 2004, 314, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Soto-Guzman, A.; Tobledo, T.; Lopez-Perez, M.; Salazar, E.P. Oleic acid induces ERK1/2 activation and AP-1 DNA binding activity through a mechanism involving Src kinase and EGFR transactivation in breast cancer cells. Mol. Cell. Endocrinol. 2008, 294, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tito, N.; Robledo, T.; Salazar, E.P. Arachidonic acid promotes FAK activation and migration in MDA-MB-231 breast cancer cells. Exp. Cell. Res. 2008, 314, 3340–3355. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Lee, Y.S.; Mayoral, R.; Oh, D.Y.; Webster, N.J.; Sears, D.D.; Olefsky, J.M.; Ellies, L.G. ω-3 Fatty acids reduce obesity-induced tumor progression independent of GPR120 in a mouse model of postmenopausal breast cancer. Oncogene 2014, 34, 3504–3513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Knoepp, S.M.; Sansbury, H.M.; Han, S.; Ku, H.; Xie, Y.; Hallman, M.; Meier, K.E. Differential expression of FAK and Pyk2 in phorbol ester-sensitive and -resistant EL4 thymoma cells. Clin. Expt. Metastasis 2011, 28, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Papupinthu, N.; Lee, H.Y.; Mills, G.B. Lysophosphatidic acid production and action: Critical new players in breast cancer initiation and progression. Br. J. Cancer 2010, 102, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed]
- O’Kelly, J.; Chung, A.; Lemp, N.; Chumakova, K.; Yin, D.; Wang, H.J.; Said, J.; Gui, D.; Miller, C.W.; Karlan, B.Y.; et al. Functional domains of CCN1 (Cyr61) regulate breast cancer progression. Int. J. Oncol. 2008, 33, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nature Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Serre, C.M.; Gres, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clezardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest. 2004, 114, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Towers, L.N.; O’Connor, K.L. LPA2 (EDG4) mediates Rho-dependent chemotaxis with lower efficacy than LPA1 (EDG2) in breast carcinoma cells. Am. J. Physiol. Cell. Physiol. 2007, 292, C1947–C1933. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Alemayehu, M.; Aziziyeh, A.; Pape, C.; Pampilio, M.; Postovit, L.M.; Mills, G.B.; Babway, A.V.; Bhattacharya, M. Beta-arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol. Cancer Res. 2009, 7, 1064–1077. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Alemayehu, M.; Dragan, M.; Pape, C.; Siddiqui, I.; Sacks, D.B.; di Guglielmo, G.M.; Babway, A.V.; Bhattacharya, M. β-Arrestin2 regulates lysophosphatidic acid-induced human breast tumor cell migration and invasion via Rap1 and IQGAP1. PLoS ONE 2013, 8, e56174. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Cai, H.; Duan, X.; Yang, Y.; Li, M.; Wu, J.; Zhang, X.; Wang, J. Aberrant expression and potential therapeutic target of lysophosphatidic acid receptor 3 in triple-negative breast cancers. Clin. Exp. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Sata, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; et al. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Liu, Z.; Meier, K.E. Cross-talk between lysophosphatidic acid receptor a, free fatty acid receptor 4, and epidermal growth factor receptor in human prostate cancer cells. 2015; Submitted. [Google Scholar]
- Boucharaba, A.; Guillet, B.; Menaa, F.; Hneino, M.; van Wijnen, A.J.; Clezardin, P.; Peyruchaud, O. Bioactive lipids lysophosphatidic acid and sphingosine 1-phosphate mediate breast cancer cell biological functions through distinct mechanisms. Oncol. Res. 2009, 18, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Sun, C.; Hu, Z.; Yang, Y.; Zhu, Y.; Zheng, D.; Gu, L.; Lu, X. Lysophosphatidic acid induces MDA-MB-231 breast cancer cells migration through activation of PI3K/PAK1/ERK signaling. PLoS ONE 2010, 5, e15940. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Nguyen, D.; Allen, L.D.; Eddy, J.; Dreau, D. Progranulin stimulated by LPA promotes the migration of aggressive breast cancer cells. Cell. Commun. Adhes. 2010, 18, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Duan, X.; Cai, H.; Liu, X.; Yang, Y.; Li, M.; Zhang, X.; Wang, J. Curcumin inhibits LPA-induced invasion by attenuating RhoA/ROCK/MMPs pathway in MCF7 breast cancer cells. Clin. Exp. Med. 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, X.; Gajewiak, J.; Tsukahara, R.; Fujiwara, Y.; Liu, J.; Fells, J.I.; Perygin, D.; Parrill, A.L.; Tigyi, G.; et al. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression. Cancer Res. 2009, 69, 5441–5449. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Daaka, Y. Mitogenic action of LPA in prostate. Biochim. Biophys. Acta. 2002, 1582, 265–269. [Google Scholar] [CrossRef]
- Marshall, J.C.A.; Collins, J.W.; Nakayama, J.; Horak, C.E.; Liewehr, D.J.; Steinberg, S.M.; Albaugh, M.; Vidal-Vanaclocha, F.; Palmieri, D.; Marbier, M.; et al. Effects of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J. Natl. Cancer Inst. 2012, 104, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Kue, P.F.; Taub, J.S.; Harrington, L.B.; Polakiewicz, R.D.; Ullrich, A.; Daaka, Y. Lysophosphatidic acid-regulated mitogenic ERK signaling in androgen-insensitive prostate cancer PC-3 cells. Int. J. Cancer 2002, 102, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Bektas, M.; Payne, S.G.; Liu, H.; Goparaju, S.; Milstien, S.; Spiegel, S. A novel acylglycerol kinase that produces lysophophatidic modulates cross talk with EGFR in prostate cancer cells. J. Cell. Biol. 2005, 169, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Snider, A.J.; Zhang, Z.; Xie, Y.; Meier, K.E. Epidermal growth factor increases lysophosphatidic acid production in human ovarian cancer cells: Roles for phospholipase D2 and receptor transactivation. Am. J. Physiol. Cell. Physiol. 2009, 298, C163–C170. [Google Scholar] [CrossRef] [PubMed]
- Mausbacher, N.; Schreiber, T.B.; Daub, H. Glycoprotein capture and quantitative phosphoproteomics indicate coordinated regulation of cell migration upon lysophosphatidic acid stimulation. Mol. Cell. Proteom. 2010, 9, 2337–2353. [Google Scholar] [CrossRef] [PubMed]
- Colin-Santana, C.C.; Avendano-Vazquez, S.E.; Alcantara-Hernandez, R.; Garcia-Sainz, J.A. EGF and angiotensin II modulate lysophosphatidic acid LPA1 receptor function and phosphorylation state. Biochim. Biophys. Acta 2011, 1810, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hopkins, M.M.; Zhang, Z.; Liu, Z.; Meier, K.E. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. J. Clin. Med. 2016, 5, 16. https://doi.org/10.3390/jcm5020016
Hopkins MM, Zhang Z, Liu Z, Meier KE. Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. Journal of Clinical Medicine. 2016; 5(2):16. https://doi.org/10.3390/jcm5020016
Chicago/Turabian StyleHopkins, Mandi M., Zhihong Zhang, Ze Liu, and Kathryn E. Meier. 2016. "Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells" Journal of Clinical Medicine 5, no. 2: 16. https://doi.org/10.3390/jcm5020016
APA StyleHopkins, M. M., Zhang, Z., Liu, Z., & Meier, K. E. (2016). Eicosopentaneoic Acid and Other Free Fatty Acid Receptor Agonists Inhibit Lysophosphatidic Acid- and Epidermal Growth Factor-Induced Proliferation of Human Breast Cancer Cells. Journal of Clinical Medicine, 5(2), 16. https://doi.org/10.3390/jcm5020016