
The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease

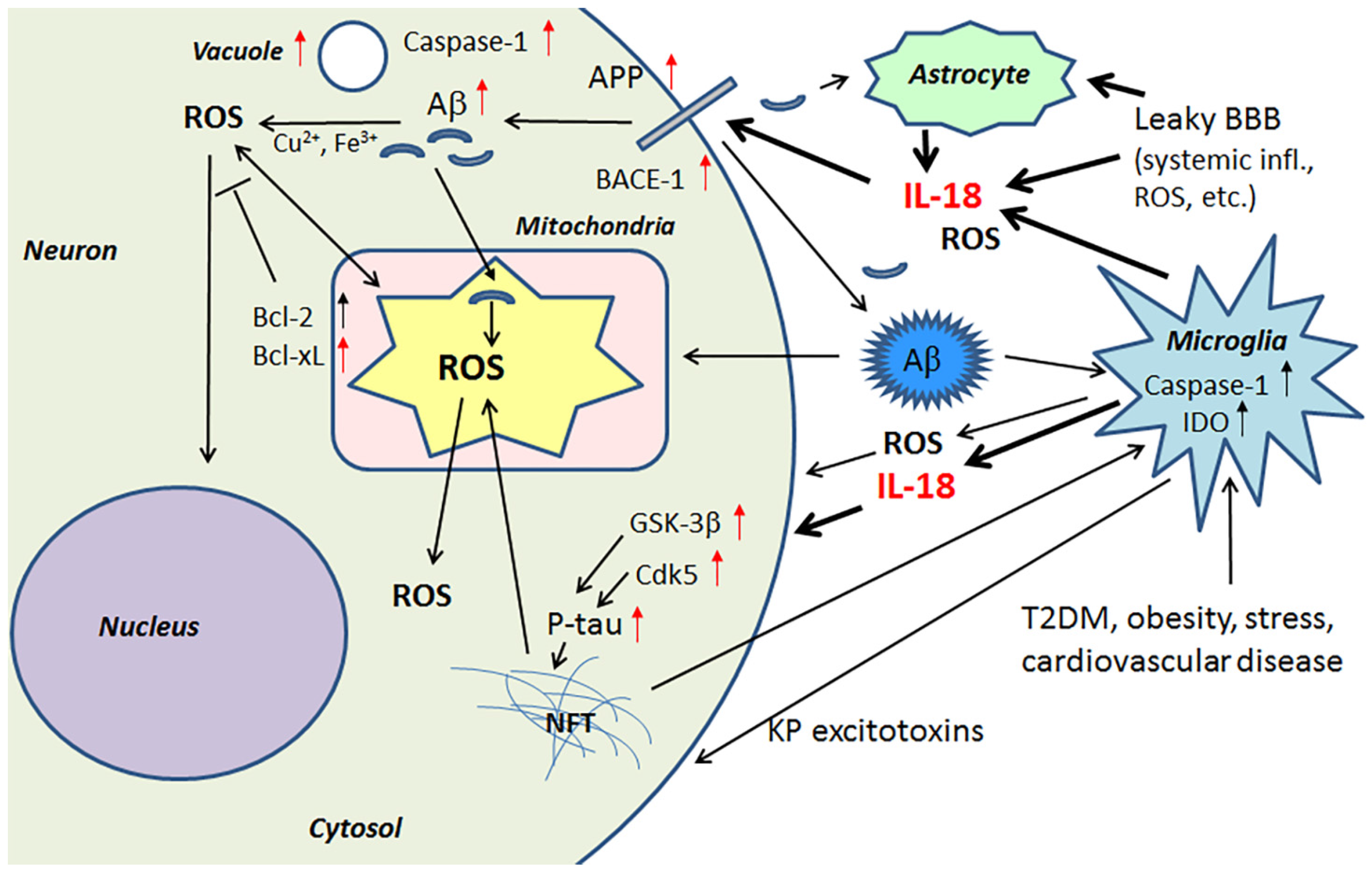{kind=link}
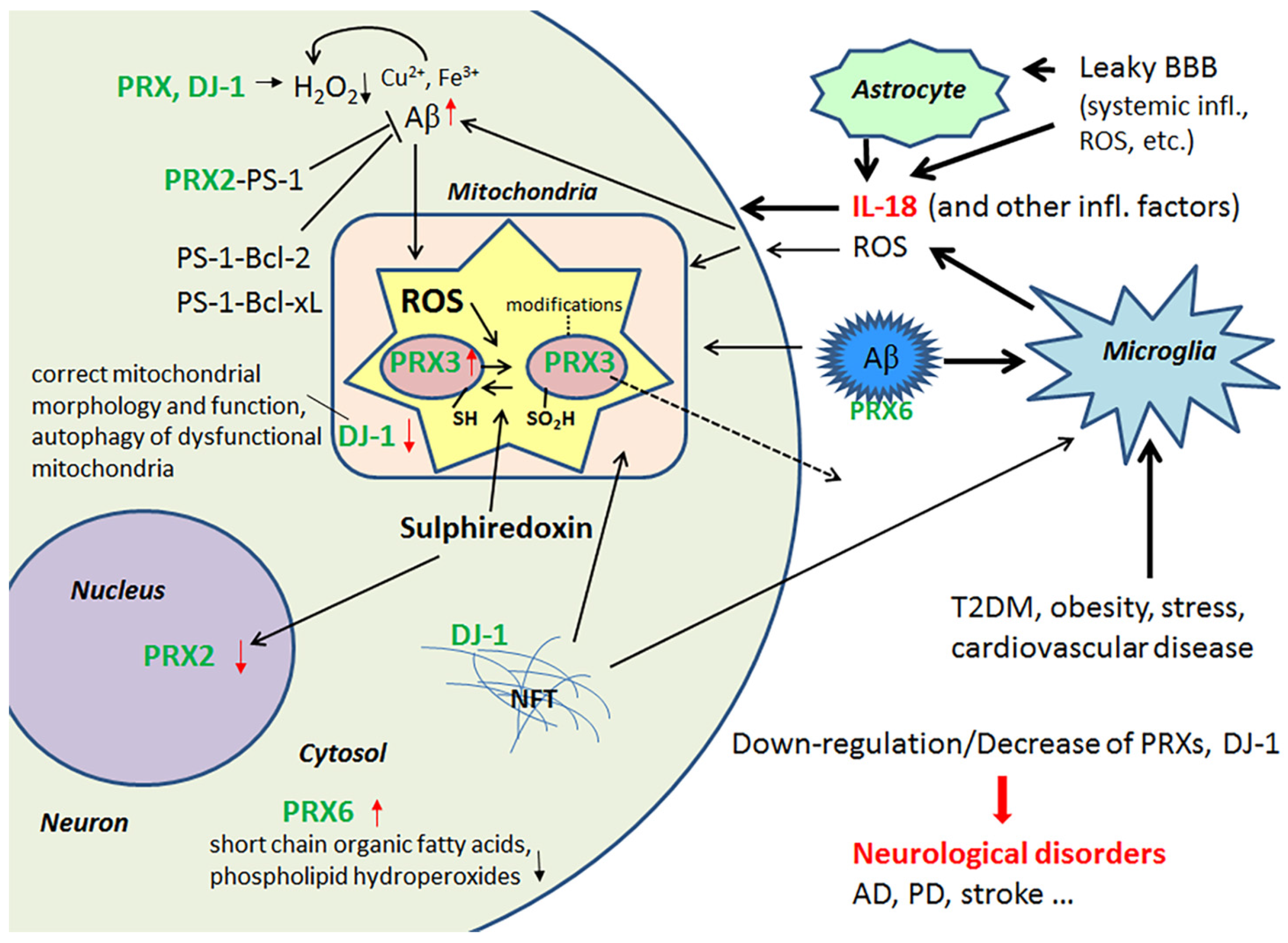{kind=link}
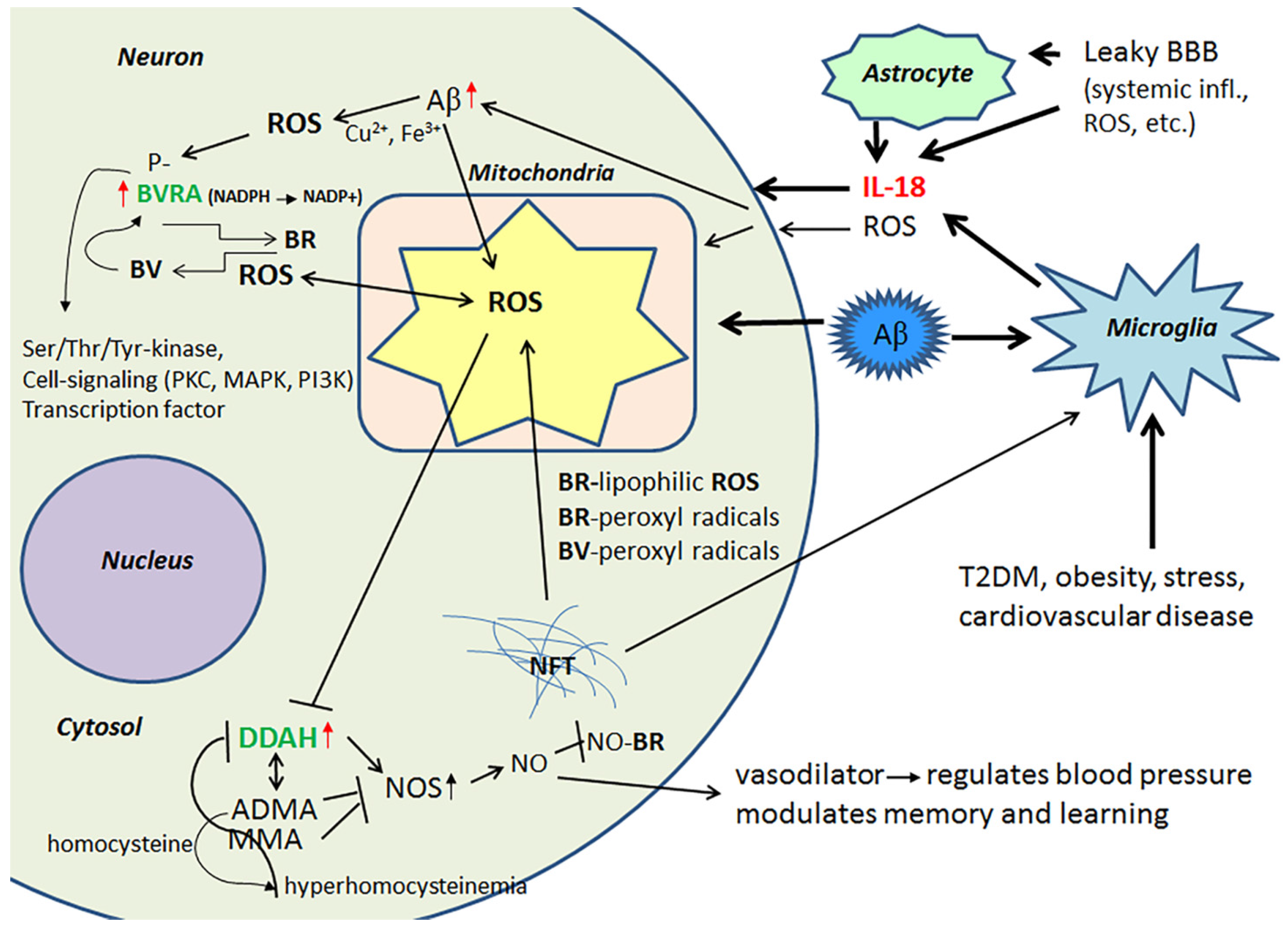{kind=link}
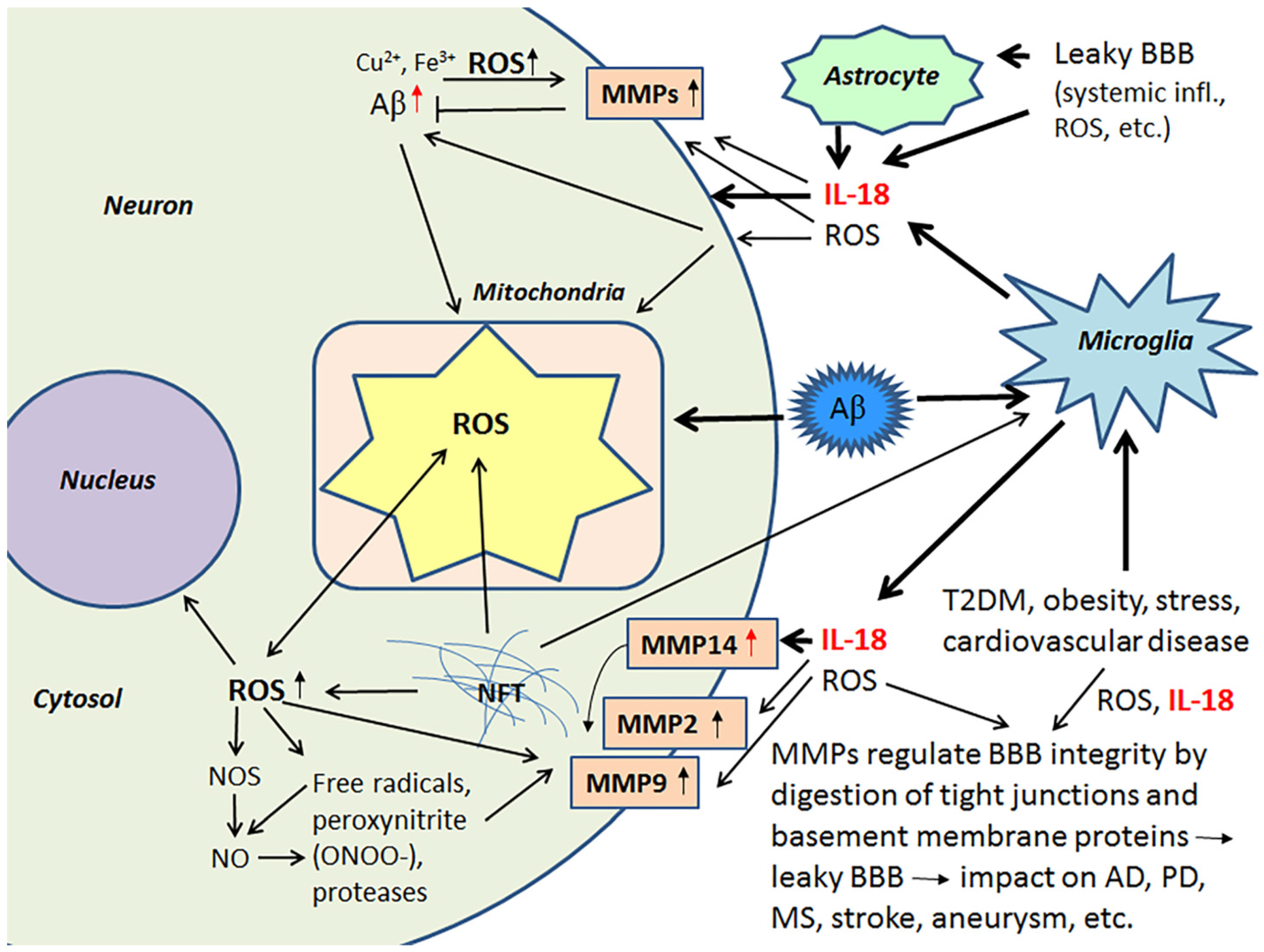{kind=link}
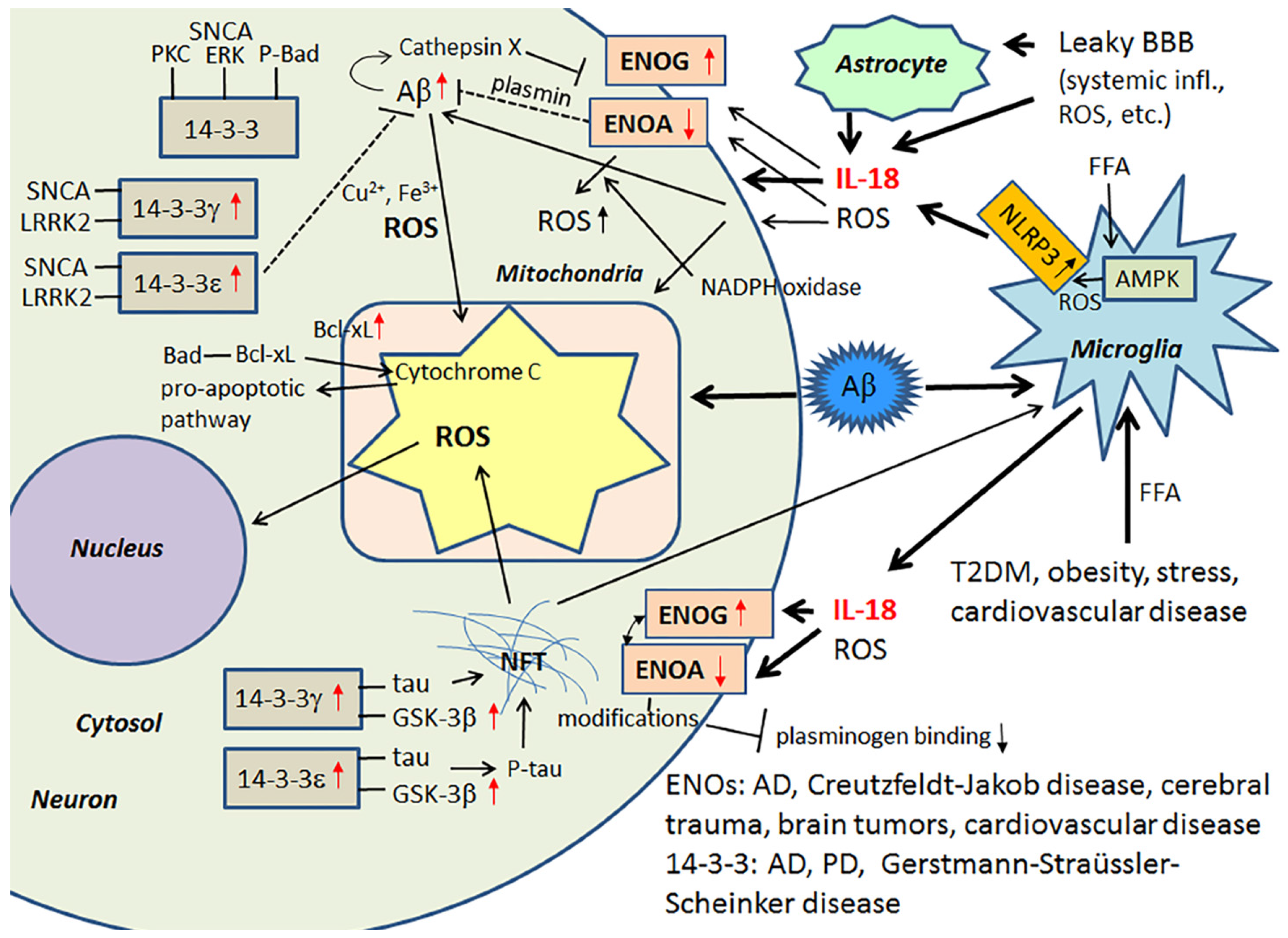{kind=link}
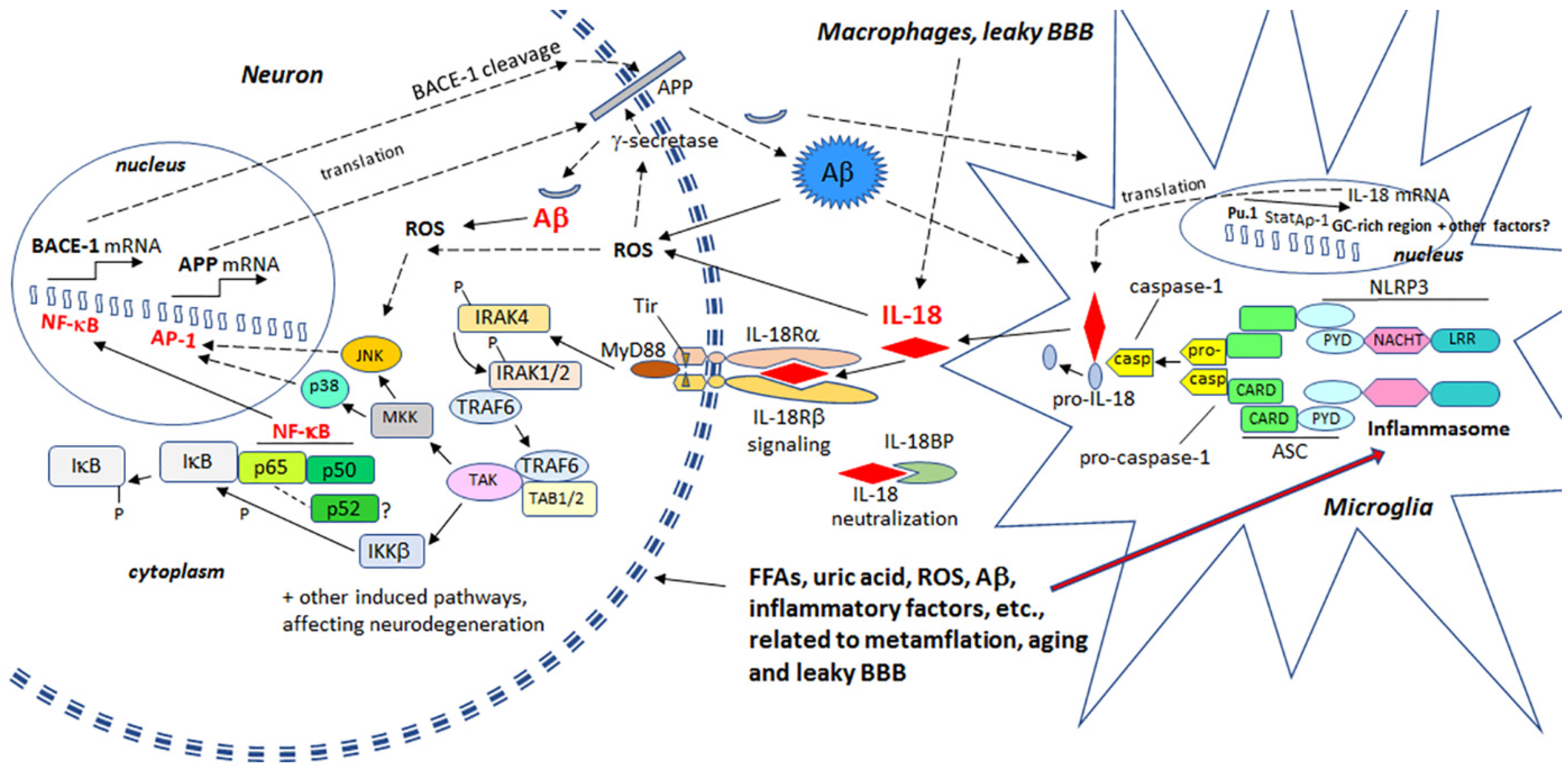{kind=link}
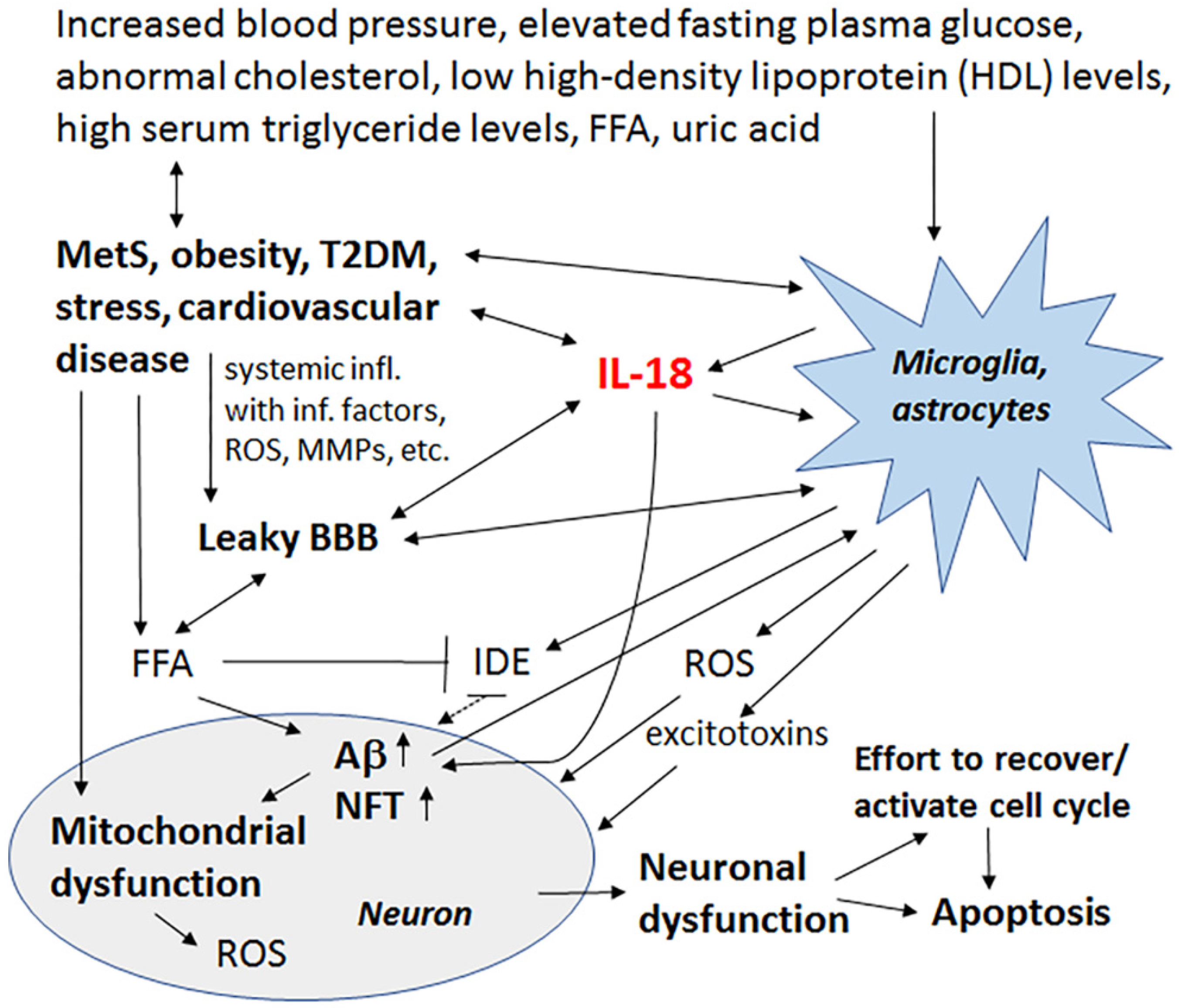{kind=link}
Abstract
:1. Introduction
2. Proinflammatory Cytokine IL-18 and Hallmarks of Alzheimer’s Disease
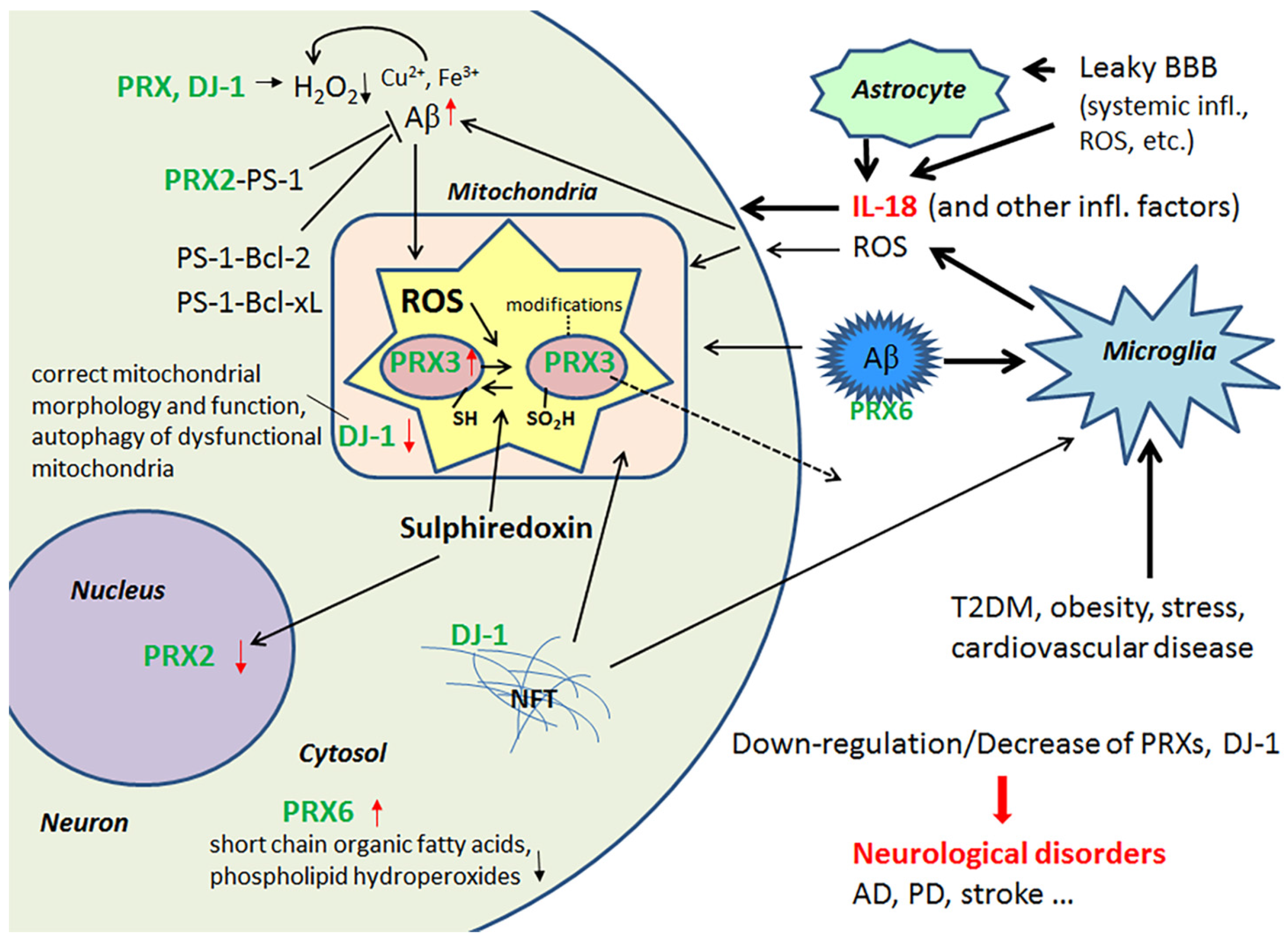
2.1. Peroxiredoxins and Protein Deglycase DJ-1 and Their Role as Antioxidants in the Brain
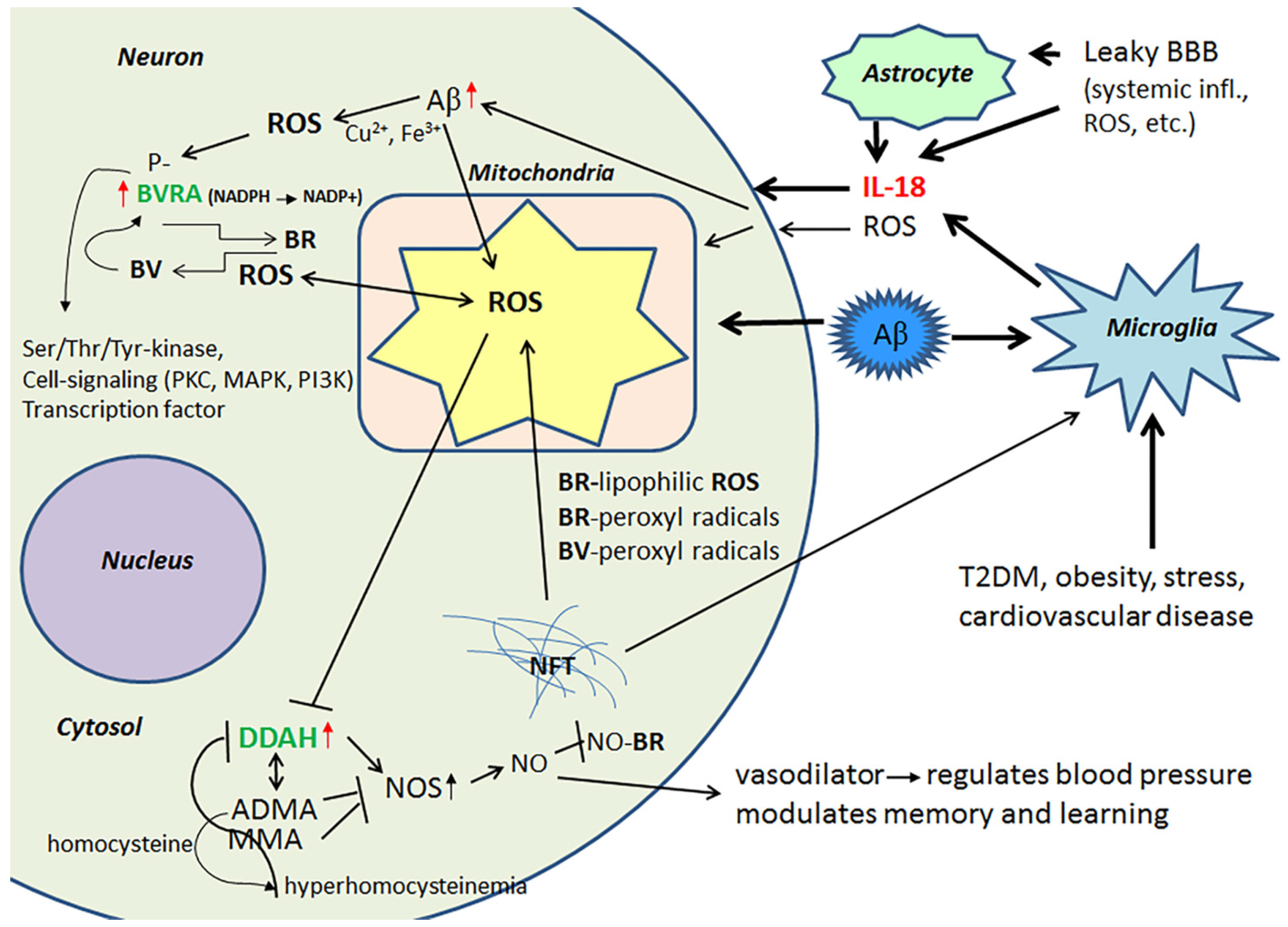
2.2. Biliverdin/Bilirubin-System, Dimethylarginine Dimethylaminohydrolase 2 and Nitrosative Stress
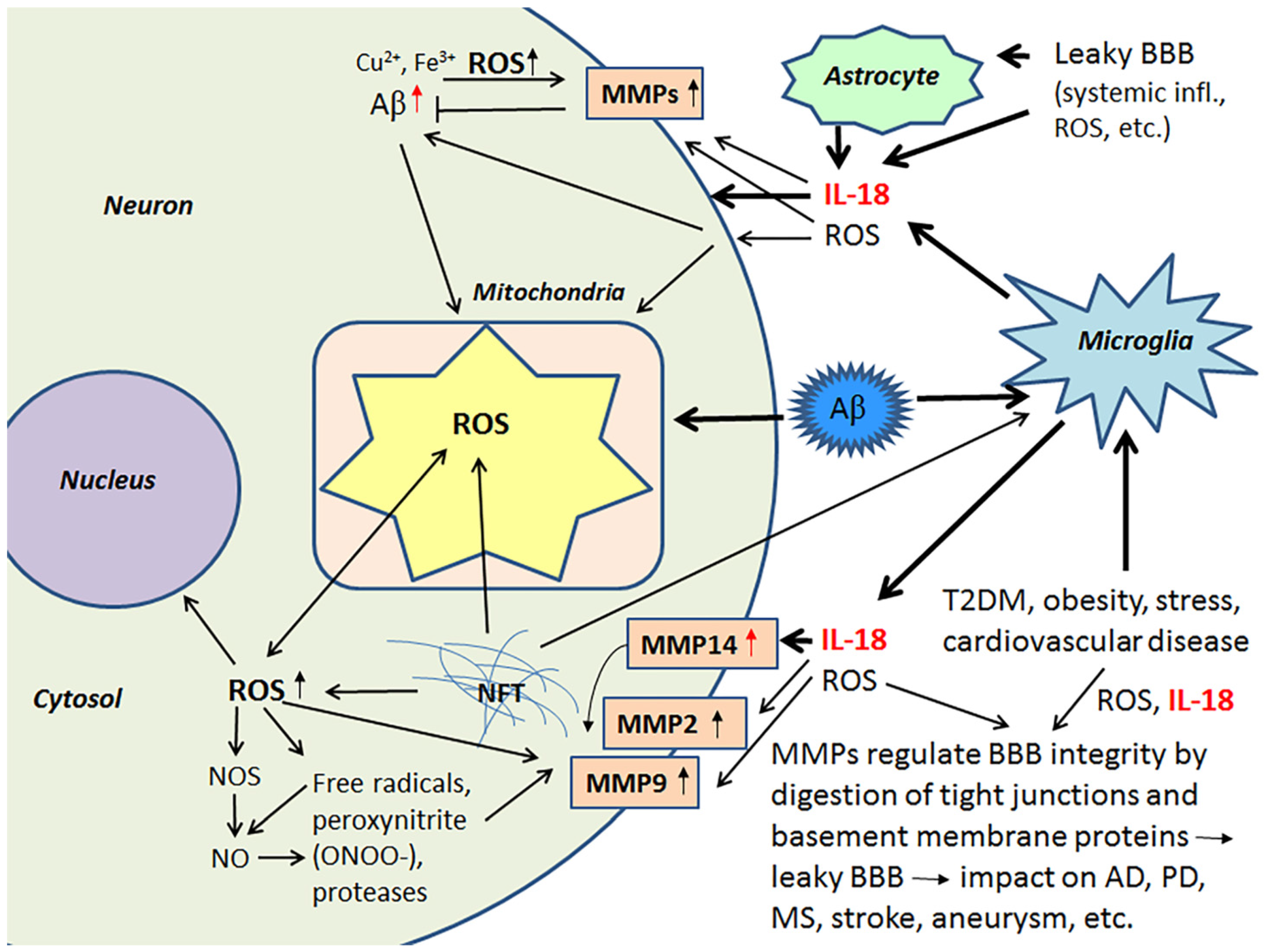
2.3. IL-18, Oxidative and Nitrosative Stress and Matrix Metalloproteinases as Regulators of the Blood-Brain Barrier
3. Metabolic Syndrome
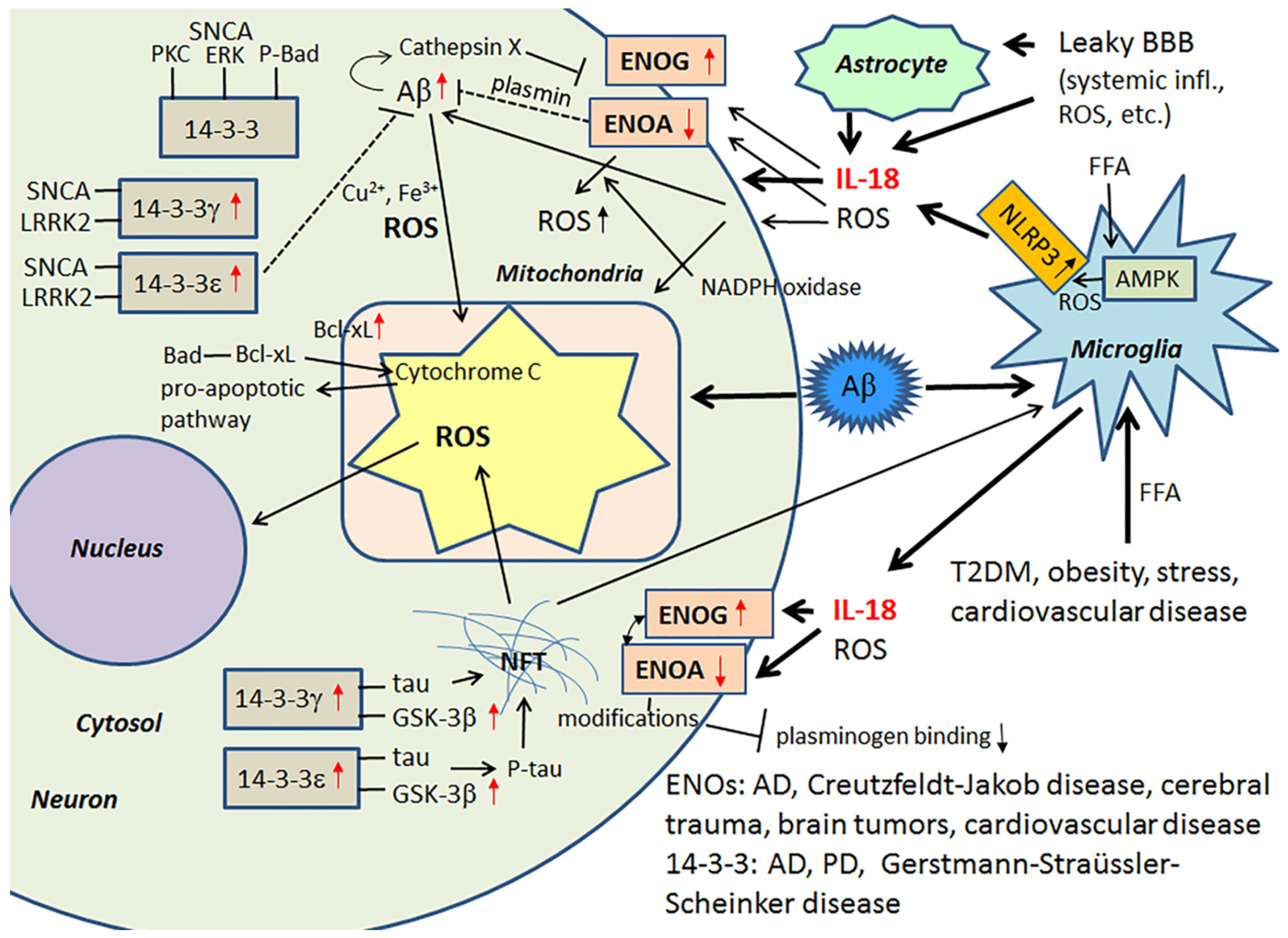
3.1. IL-18, Enolases and Multiple Functioning 14-3-3
4. The Effect of Lifestyle and Diet on the Genesis/Progression of Alzheimer’s Disease
4.1. Lipid Rafts and Their Composition
5. Summary, Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Krapfenbauer, K.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003, 967, 152–160. [Google Scholar] [CrossRef]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Essick, E.E.; Sam, F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell Longev. 2010, 3, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, L.F.; Titov, V.N. Lipid peroxidation in relation to ageing and the role of endogenous aldehydes in diabetes and other age-related diseases. Ageing Res. Rev. 2010, 9, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Bendlin, B.B.; Carlsson, C.M.; Gleason, C.E.; Johnson, S.C.; Sodhi, A.; Gallagher, C.L.; Puglielli, L.; Engelman, C.D.; Ries, M.L.; Xu, G.; et al. Midlife predictors of Alzheimer’s disease. Maturitas 2010, 65, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Hamer, M.; Sabia, S.; Batty, G.D.; Shipley, M.J.; Tabák, A.G.; Singh-Manoux, A.; Kivimaki, M. Physical activity and inflammatory markers over 10 years: Follow-up in men and women from the Whitehall II cohort study. Circulation 2012, 126, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenín, J. Alzheimer’s disease: Redox dysregulation as a common denominator for diverse pathogenic mechanisms. Antioxid. Redox Signal. 2012, 16, 974–1031. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Endo, T. Survival responses to oxidative stress and aging. Geriatr. Gerontol. Int. 2010, 10 (Suppl. 1), S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Klann, E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid. Redox Signal. 2007, 9, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.M.; Lawrence, D.A.; Mondal, T.K.; Seegal, R.F. Reduced glutathione is highly expressed in white matter and neurons in the unperturbed mouse brain—Implications for oxidative stress associated with neurodegeneration. Brain Res. 2009, 1276, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, C.A.; Egaña, J.T.; Núñez, M.T.; Maccioni, R.B.; González-Billault, C. Oxidative stress promotes tau dephosphorylation in neuronal cells: The roles of cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Coombes, E.; Jiang, J.; Chu, X.P.; Inoue, K.; Seeds, J.; Branigan, D.; Simon, R.P.; Xiong, Z.G. Pathophysiological relevant levels of hydrogen peroxide induces glutamate-independent neurodegeneration that involves activation of transient receptor potential melastatin 7 channels. Antioxid. Redox Signal. 2011, 14, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Ojala, J.O.; Sutinen, E.M.; Salminen, A.; Pirttilä, T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J. Neuroimmunol. 2008, 205, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Deng, W.; Wang, Z.; Ning, M.; Zhang, W.; Zhou, Y.; Lo, E.H.; Xing, C. Differential subnetwork of chemokines/cytokines in human, mouse, and rat brain cells after oxygen-glucose deprivation. J. Cereb. Blood Flow Metab. 2016, 37, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Herance, R.; Rovira, S.; Hernández-Mijares, A.; Victor, V.M. Mitochondrial dysfunction and antioxidant therapy in sepsis. Infect. Disord. Drug Targets 2012, 12, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.; Tharakan, B. Vascular hyperpermeability and aging. Aging Dis. 2014, 5, 114–125. [Google Scholar] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell. Mol. Life Sci. 2012, 69, 2999–3013. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttilä, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Guzmán-Martínez, L.; Cerda-Troncoso, C.; Farías, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Rossner, S.; Sastre, M.; Bourne, K.; Lichtenthaler, S.F. Transcriptional and translational regulation of BACE1 expression—Implications for Alzheimer’s disease. Prog. Neurobiol. 2006, 79, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys Acta 2014, 1842, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Opazo, C.; Huang, X.; Cherny, R.A.; Moir, R.D.; Roher, A.E.; White, A.R.; Cappai, R.; Masters, C.L.; Tanzi, R.E.; Inestrosa, N.C.; et al. Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J. Biol. Chem. 2002, 277, 40302–40308. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Yang, S.G.; Liu, W.; Zhang, Y.X.; Xu, P.X.; Wang, T.; Ling, T.J.; Liu, R.T. Alpha-tocopherol quinine ameliorates spatial memory deficits by reducing beta-amyloid oligomers, neuroinflammation and oxidative stress in transgenic mice with Alzheimer’s disease. Behav. Brain Res. 2016, 296, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Islam, M.N.; Ali, M.Y.; Kim, Y.M.; Park, H.J.; Sohn, H.S.; Jung, H.A. The effects of C-glycosylation of luteolin on its antioxidant, anti-Alzheimer’s disease, anti-diabetic, and anti-inflammatory activities. Arch. Pharm. Res. 2014, 37, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Mitochondrial glutathione: Features, regulation and role in disease. Biochim. Biophys. Acta 2013, 1830, 3317–3328. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Aso, Y.; Okumura, K.; Takebayashi, K.; Wakabayashi, S.; Inukai, T. Relationships of plasma interleukin-18 concentrations to hyperhomocysteinemia and carotid intimal-media wall thickness in patients with type 2 diabetes. Diabetes Care 2003, 26, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Pontillo, A.; Ciotola, M.; Di Palo, C.; Grella, E.; Nicoletti, G.; Giugliano, D. Weight loss reduces interleukin-18 levels in obese women. J. Clin. Endocrinol. Metab. 2002, 87, 3864–3866. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Sugama, S.; Conti, B. Interleukin-18 and stress. Brain Res. Rev. 2008, 58, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, T.; Nagata, T.; Yang, D.; Nishizaki, T. Interleukin-18 regulates motor activity, anxiety and spatial learning without affecting synaptic plasticity. Behav. Brain Res. 2010, 206, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Haastrup, E.; Bukh, J.D.; Bock, C.; Vinberg, M.; Thørner, L.W.; Hansen, T.; Werge, T.; Kessing, L.V.; Ullum, H. Promoter variants in IL18 are associated with onset of depression in patients previously exposed to stressful-life events. J. Affect. Disord. 2012, 136, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Diniz, B.S.; Butters, M.A.; Albert, S.M.; Dew, M.A.; Reynolds, C.F., 3rd. Late-life depression and risk of vascular dementia and Alzheimer’s disease: Systematic review and meta-analysis of community-based cohort studies. Br. J. Psychiatry 2013, 202, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflamm. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Sutinen, E.M.; Korolainen, M.A.; Häyrinen, J.; Alafuzoff, I.; Petratos, S.; Salminen, A.; Soininen, H.; Pirttilä, T.; Ojala, J.O. Interleukin-18 alters protein expressions of neurodegenerative diseases-linked proteins in human SH-SY5Y neuron-like cells. Front. Cell Neurosci. 2014, 8, 214. [Google Scholar] [CrossRef] [PubMed]
- Gonul, Y.; Kazandi, S.; Kocak, A.; Ahsen, A.; Bal, A.; Karavelioglu, A.; Hazman, O.; Turamanlar, O.; Kokulu, S.; Yuksel, S. Interleukin-18 Binding Protein Pretreatment Attenuates Kidney Injury Induced by Hepatic Ischemia Reperfusion. Am. J. Med. Sci. 2016, 352, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflamm. 2012, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Ojala, J. Alzheimer’s and seizures: Interleukin-18, indoleamine 2,3-dioxygenase and quinolinic acid. Int. J. Tryptophan Res. 2010, 3, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Luetjens, C.M.; Lankiewicz, S.; Bui, N.T.; Krohn, A.J.; Poppe, M.; Prehn, J.H. Up-regulation of Bcl-xL in response to subtoxic beta-amyloid: Role in neuronal resistance against apoptotic and oxidative injury. Neuroscience 2001, 102, 139–150. [Google Scholar] [CrossRef]
- Liste, I.; García-García, E.; Bueno, C.; Martínez-Serrano, A. Bcl-XL modulates the differentiation of immortalized human neural stem cells. Cell Death Differ. 2007, 14, 1880–1892. [Google Scholar] [CrossRef] [PubMed]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins-Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Alberici, A.; Moratto, D.; Benussi, L.; Gasparini, L.; Ghidoni, R.; Gatta, L.B.; Finazzi, D.; Frisoni, G.B.; Trabucchi, M.; Growdon, J.H.; et al. Presenilin 1 protein directly interacts with Bcl-2. J. Biol. Chem. 1999, 274, 30764–30769. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, W.; Easton, R.; Ray, J.W.; Lampe, P.; Jiang, Z.; Brunkan, A.L.; Goate, A.; Johnson, E.M.; Wu, J.Y. Presenilin-1 protects against neuronal apoptosis caused by its interacting protein PAG. Neurobiol. Dis. 2002, 9, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Musicco, C.; Capelli, V.; Pesce, V.; Timperio, A.M.; Calvani, M.; Mosconi, L.; Zolla, L.; Cantatore, P.; Gadaleta, M.N. Accumulation of overoxidized Peroxiredoxin III in aged rat liver mitochondria. Biochim. Biophys. Acta 2009, 1787, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Power, J.H.T.; Asad, S.; Chataway, T.K.; Chegini, F.; Manavis, J.; Temlett, J.A.; Jensen, P.H.; Blumbergs, P.C.; Gai, W.-P. Peroxiredoxin 6 in human brain: Molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol. 2008, 115, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.Z.; Li, D.Q.; Hou, Y.F.; Wu, J.; Lu, J.S.; Di, G.H.; Jin, W.; Ou, Z.L.; Shen, Z.Z.; Shao, Z.M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, R76. [Google Scholar] [CrossRef] [PubMed]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [PubMed]
- Baramova, E.N.; Bajou, K.; Remacle, A.; L’Hoir, C.; Krell, H.W.; Weidle, U.H.; Noel, A.; Foidart, J.M. Involvement of PA/plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Lett. 1997, 405, 157–162. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar] [CrossRef] [PubMed]
- Baulac, S.; Lu, H.; Strahle, J.; Yang, T.; Goldberg, M.S.; Shen, J.; Schlossmacher, M.G.; Lemere, C.A.; Lu, Q.; Xia, W. Increased DJ-1 expression under oxidative stress and in Alzheimer’s disease brains. Mol. Neurodegener. 2009, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, J.; Chen, S.; Wang, Y.; Cao, L.; Zhang, Y.; Kang, W.; Li, H.; Gui, Y.; Chen, S.; et al. DJ-1 modulates the expression of Cu/Zn-superoxide dismutase-1 through the Erk1/2-Elk1 pathway in neuroprotection. Ann. Neurol. 2011, 70, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.; et al. DJ-1 colocalizes with tau inclusions: A link between parkinsonism and dementia. Ann. Neurol. 2004, 55, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Jain, R.; Eberhard, D.; Eglinger, J.; Bugliani, M.; Piemonti, L.; Marchetti, P.; Lammert, E. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J. Mol. Cell Biol. 2012, 4, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Park, S.C. Physiological antioxidative network of the bilirubin system in aging and age-related diseases. Front. Pharmacol. 2012, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Hortmann, M.; Oelze, M.; Opitz, B.; Steven, S.; Schell, R.; Knorr, M.; Karbach, S.; Schuhmacher, S.; Wenzel, P.; et al. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell Cardiol. 2010, 49, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Daiber, A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Trombino, S.; Cassano, R.; Sgambato, A.; De Paola, B.; Di Stasio, E.; Picci, N.; Preziosi, P.; Mancuso, C. Characterization of the S-denitrosylating activity of bilirubin. J. Cell Mol. Med. 2009, 13, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase—A protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 2011, 1812, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Otterbein, L.E. Go green: The anti-inflammatory effects of biliverdin reductase. Front. Pharmacol. 2012, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Marmarosh, N.; Shen, J.; Torno, M.D.; Kravets, A.; Hu, Z.; Maines, M.D. Human biliverdin reductase: A member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 7109–7114. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E.; Tudor, C.; Maines, M.D. Biliverdin reductase: More than a namesake—The reductase, its peptide fragments, and biliverdin regulate activity of the three classes of protein kinase C. Front. Pharmacol. 2012, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. Homocysteine increases the production of asymmetric dimethylarginine in cultured neurons. J. Neurosci. Res. 2004, 77, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Pope, A.J.; Karuppiah, K.; Cardounel, A.J. Role of the PRMT-DDAH-ADMA axis in the regulation of endothelial nitric oxide production. Pharmacol. Res. 2009, 60, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, L.R.; Bachetti, T.; Leiper, J.; Zachary, I.; Chenm, L.; Renné, T.; Wojciak-Stothard, B. The ADMA/DDAH pathway regulates VEGF-mediated angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, H.; Rodionov, R.N.; Lynch, C.; Cooke, J.P.; Arning, E.; Bottiglieri, T.; Lentz, S.R.; Faraci, F.M. Overexpression of dimethylarginine dimethylaminohydrolase inhibits asymmetric dimethylarginine-induced endothelial dysfunction in the cerebral circulation. Stroke 2008, 39, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Stühlinger, M.C.; Stanger, O. Asymmetric dimethyl-L-arginine (ADMA): A possible link between homocyst(e)ine and endothelial dysfunction. Curr. Drug Metab. 2005, 6, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and neurologic disorders: A review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE 2011, 6, e20599. [Google Scholar] [CrossRef]
- Lischper, M.; Beuck, S.; Thanabalasundaram, G.; Pieper, C.; Galla, H.J. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010, 1326, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Cauwe, B.; Van den Steen, P.E.; Opdenakker, G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 113–185. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.J.; Butler, G.S.; Rodriguez, D.; Overall, C.M. Matrix metalloproteinase proteomics: Substrates, targets, and therapy. Curr. Opin. Cell Biol. 2009, 21, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [PubMed]
- Pun, P.B.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Gasche, Y.; Grzeschik, S.; Copin, J.C.; Maier, C.M.; Chan, P.H. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: Role of matrix metalloproteinase-9 in early blood-brain barrier disruption? J. Neurosci. 2003, 23, 8733–8742. [Google Scholar] [PubMed]
- Ridnour, L.A.; Dhanapal, S.; Hoos, M.; Wilson, J.; Lee, J.; Cheng, R.Y.; Brueggemann, E.E.; Hines, H.B.; Wilcock, D.M.; Vitek, M.P.; et al. Nitric oxide-mediated regulation of β-amyloid clearance via alterations of MMP-9/TIMP-1. J. Neurochem. 2012, 123, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Migita, K.; Izumi, Y.; Nakao, K.; Ida, H.; Kawakami, A.; Abiru, S.; Ishibashi, H.; Eguchi, K.; Ishii, N. The role of IL-18 in the modulation of matrix metalloproteinases and migration of human natural killer (NK) cells. FEBS Lett. 2004, 569, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, B.; Mummidi, S.; Mahimainathan, L.; Patel, D.N.; Bailey, S.R.; Imam, S.Z.; Greene, W.C.; Valente, A.J. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J. Biol. Chem. 2006, 281, 15099–15109. [Google Scholar] [CrossRef] [PubMed]
- Lehti, K.; Lohi, J.; Valtanen, H.; Keski-Oja, J. Proteolytic processing of membrane-type-1 matrix metalloproteinase is associated with gelatinase A activation at the cell surface. Biochem. J. 1998, 334 Pt 2, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Takamura, A.; Ito, N.; Maru, Y.; Sato, H.; Suenaga, N.; Aoki, T.; Seiki, M. Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J. 2001, 20, 4782–4793. [Google Scholar] [CrossRef] [PubMed]
- Merlo, S.; Sortino, M.A. Estrogen activates matrix metalloproteinases-2 and -9 to increase beta amyloid degradation. Mol. Cell Neurosci. 2012, 49, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.C.; Van Nostrand, W.E. Degradation of soluble and fibrillar amyloid beta-protein by matrix metalloproteinase (MT1-MMP) in vitro. Biochemistry 2010, 49, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Weekman, E.M.; Wilcock, D.M. Matrix Metalloproteinase in Blood-Brain Barrier Breakdown in Dementia. J. Alzheimers Dis. 2015, 49, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Reddy, P.H. Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. J. Alzheimers Dis. 2016, 54, 427–443. [Google Scholar] [CrossRef] [PubMed]
- McColl, B.W.; Rose, N.; Robson, F.H.; Rothwell, N.J.; Lawrence, C.B. Increased brain microvascular MMP-9 and incidence of haemorrhagic transformation in obese mice after experimental stroke. J. Cereb. Blood Flow Metab. 2010, 30, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zhang, J.; Feng, C.; Xiong, L.; Zuo, Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in mice. Cardiovasc. Res. 2014, 103, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Dik, M.G.; Jonker, C.; Comijs, H.C.; Deeg, D.J.; Kok, A.; Yaffe, K.; Penninx, B.W. Contribution of metabolic syndrome components to cognition in older individuals. Diabetes Care 2007, 30, 2655–2660. [Google Scholar] [CrossRef] [PubMed]
- Campos-Peña, V.; Toral-Rios, D.; Becerril-Pérez, F.; Sánchez-Torres, C.; Delgado-Namorado, Y.; Torres-Ossorio, E.; Franco-Bocanegra, D.; Carvajal, K. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: Is Aβ a Crucial Factor in Both Pathologies? Antioxid. Redox Signal. 2017, 26, 542–560. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 2001, 74, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Park, C.R.; Seeley, R.J.; Craft, S.; Woods, S.C. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol. Behav. 2000, 68, 9–14. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, AM.; Greene, M.W.; Thangiah, G.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta 2017, 1863, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Jha, N.K.; Kumar, D.; Ambasta, R.K.; Kumar, P. Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in neurodegeneration. Biochim. Biophys. Acta 2017, 1863, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Treviño, S.; Auilar-Alonso, P.; Flores Hernandez, J.A.; Brambila, E.; Guevara, J.; Flores, G.; Lopez-Lopez, G.; Muñoz-Arenas, G.; Morales-Medina, J.C.; Toxqui, V.; et al. A high calorie diet causes memory loss, metabolic syndrome and oxidative stress into hippocampus and temporal cortex of rats. Synapse 2015, 69, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Ting, J.P.; O’Neill, L.A. A role for the NLRP3 inflammasome in metabolic diseases—Did Warburg miss inflammation? Nat. Immunol. 2012, 13, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Capello, M.; Ferri-Borgogno, S.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget 2016, 7, 5598–5612. [Google Scholar] [PubMed]
- Butterfield, D.A.; Bader Lange, M.L. Multifunctional roles of enolase in Alzheimer’s disease brain: Beyond altered glucose metabolism. J. Neurochem. 2009, 111, 915–933. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Boyd-Kimball, D.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Butterfield, D.A. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J. Alzheimers Dis. 2007, 11, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Aksenov, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 2002, 82, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.B.; Di Domenico, F.; Sultana, R.; Perluigi, M.; Cini, C.; Pierce, W.M.; Butterfield, D.A. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: Implications for progression of AD. J. Proteome Res. 2009, 8, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M., Jr.; Turner, D.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J. Cell Mol. Med. 2009, 13, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Ramos, A.; Roig-Borrellas, A.; García-Melero, A.; López-Alemany, R. α-Enolase, a multifunctional protein: Its role on pathophysiological situations. J. Biomed. Biotechnol. 2012, 2012, 156795. [Google Scholar] [CrossRef] [PubMed]
- Sinniger, V.; Merton, R.E.; Fabregas, P.; Felez, J.; Longstaff, C. Regulation of tissue plasminogen activator activity by cells. Domains responsible for binding and mechanism of stimulation. J. Biol. Chem. 1999, 274, 12414–12422. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Pizzimenti, S.; Arcaro, A.; Pettazzoni, P.; Minelli, R.; D’Angelo, D.; Mamone, G.; Ferranti, P.; Toaldo, C.; Cetrangolo, G.; et al. Exposure of HL-60 human leukaemic cells to 4-hydroxynonenal promotes the formation of adduct(s) with alpha-enolase devoid of plasminogen binding activity. Biochem. J. 2009, 422, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Aksamit, A.J., Jr.; Preissner, C.M.; Homburger, H.A. Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology 2001, 57, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Obermajer, N.; Kos, J. γ-1-syntrophin mediates trafficking of γ-enolase towards the plasma membrane and enhances its neurotrophic activity. Neurosignals 2010, 18, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Obermajer, N.; Kos, J. γ-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem. J. 2012, 443, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Wendt, W.; Zhu, X.R.; Lübbert, H.; Stichel, C.C. Differential expression of cathepsin X in aging and pathological central nervous system of mice. Exp. Neurol. 2007, 204, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Obermajer, N.; Doljak, B.; Jamnik, P.; Fonović, U.P.; Kos, J. Cathepsin X cleaves the C-terminal dipeptide of alpha- and gamma-enolase and impairs survival and neuritogenesis of neuronal cells. Int. J. Biochem. Cell Biol. 2009, 41, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Aitken, A.; Otto, M. 14-3-3 proteins in neurodegeneration. Semin. Cell Dev. Biol. 2011, 22, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova, N.; Petrucelli, L.; Farrer, M.; Mehta, N.; Choi, P.; Hardy, J.; Wolozin, B. Alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 1999, 19, 5782–5791. [Google Scholar] [PubMed]
- Cohen, P.; Goedert, M. GSK3 inhibitors: Development and therapeutic potential. Nat. Rev. Drug Discov. 2004, 3, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Agarwal-Mawal, A.; Paudel, H.K. 14-3-3 binds to and mediates phosphorylation of microtubule-associated tau protein by Ser9-phosphorylated glycogen synthase kinase 3beta in the brain. J. Biol. Chem. 2004, 279, 26105–26114. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, L.; Correani, V.; Fabrizi, C.; Fumagalli, L.; Mazzanti, M.; Maras, B.; Schininà, M.E. 14-3-3ε marks the amyloid-stimulated microglia long-term activation. Proteomics 2012, 12, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Giaccone, G.; Limido, L.; Mangieri, M.; Suardi, S.; Puoti, G.; Morbin, M.; Mazzoleni, G.; Ghetti, B.; Tagliavini, F. The ε Isoform of 14-3-3 Protein Is a Component of the Prion Protein Amyloid Deposits of Gerstmann-Straüssler-Scheinker Disease. J. Neuropathol. Exp. Neurol. 2007, 66, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Zhu, X.; Babar, A.K.; Siedlak, S.L.; Yang, Q.; Ito, G.; Iwatsubo, T.; Smith, M.A.; Chen, S.G. Leucine-rich repeat kinase 2 colocalizes with alpha-synuclein in Parkinson’s disease, but not tau-containing deposits in tauopathies. Neurodegener. Dis. 2008, 5, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Q.J.; Pan, N.; Lee, S.; Zhao, Y.; Chait, B.T.; Yue, Z. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS ONE 2011, 6, e17153. [Google Scholar]
- Yacoubian, T.A.; Slone, S.R.; Harrington, A.J.; Hamamichi, S.; Schieltz, J.M.; Caldwell, K.A.; Caldwell, G.A.; Standaert, D.G. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell Death Dis. 2010, 1, e2. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.O. Melatonin attenuates the focal cerebral ischemic injury by inhibiting the dissociation of pBad from 14-3-3. J. Pineal Res. 2008, 44, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Xu, G.; Nagel, D.J.; Hua, Z.; Zhang, N.; Yin, G. A model of ischemia and reperfusion increases JNK activity, inhibits the association of BAD and 14-3-3, and induces apoptosis of rabbit spinal neurocytes. Neurosci. Lett. 2010, 473, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.S.; Cheung, W.M.; Tsai, Y.S.; Chen, Y.T.; Fong, W.H.; Tsai, H.D.; Chen, Y.C.; Liou, J.Y.; Shyue, S.K.; Chen, J.J.; et al. Ligand-activated peroxisome proliferator-activated receptor-gamma protects against ischemic cerebral infarction and neuronal apoptosis by 14-3-3 epsilon upregulation. Circulation 2009, 119, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.A.; Ferrioli, E.; Nakano, E.Y.; Litvoc, J.; Bottino, C.M. High prevalence of dementia in a community-based survey of older people from Brazil: Association with intellectual activity rather than education. J. Alzheimers Dis. 2012, 32, 307–316. [Google Scholar] [PubMed]
- Vemuri, P.; Lesnick, T.G.; Przybelski, S.A.; Knopman, D.S.; Roberts, R.O.; Lowe, V.J.; Kantarci, K.; Senjem, M.L.; Gunter, J.L.; Boeve, B.F.; et al. Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Ann. Neurol. 2012, 72, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Cass, S.P. Alzheimer’s Disease and Exercise: A Literature Review. Curr. Sports Med. Rep. 2017, 16, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Castellano, C.A.; Paquet, N.; Dionne, I.J.; Imbeault, H.; Langlois, F.; Croteau, E.; Tremblay, S.; Fortier, M.; Matte, J.J.; Lacombe, G.; et al. A 3-Month Aerobic Training Program Improves Brain Energy Metabolism in Mild Alzheimer’s Disease: Preliminary Results from a Neuroimaging Study. J. Alzheimers Dis. 2017, 56, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic exercise for Alzheimer’s disease: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, D.; Slørdahl, S.A.; Wisløff, U. Effect of exercise training on inflammation status among people with metabolic syndrome. Metab. Syndr. Relat. Disord. 2012, 10, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Raefsky, S.M.; Mattson, M.P. Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Kihlström, M.; Ojala, J.; Salminen, A. Decreased level of cardiac antioxidants in endurance-trained rats. Acta Physiol. Scand. 1989, 135, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Haboubi, N. Assessment and management of nutrition in older people and its importance to health. Clin. Interv. Aging 2010, 5, 207–216. [Google Scholar] [PubMed]
- Lövdén, M.; Xu, W.; Wang, H.-X. Lifestyle change and the prevention of cognitive decline and dementia: What is the evidence? Curr. Opin. Psychiatry 2013, 26, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Marziano, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Nutrition and AGE-ing: Focusing on Alzheimer’s Disease. Oxid. Med. Cell Longev. 2017, 2017, 7039816. [Google Scholar] [CrossRef] [PubMed]
- Reedy, J.; Krebs-Smith, S.M.; Miller, P.E.; Liese, A.D.; Kahle, L.L.; Park, Y.; Subar, A.F. Higher diet quality is associated with decreased risk of all-cause, cardiovascular disease, and cancer mortality among older adults. J. Nutr. 2014, 144, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.; Imrhan, V.; Marotta, F.; Juma, S.; Vijayagopal, P. Lifestyle and advanced glycation end products (AGEs) burden: Its relevance to healthy aging. Aging Dis. 2014, 5, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Dias, I.H.; Mistry, J.; Fell, S.; Reis, A.; Spickett, C.M.; Polidori, M.C.; Lip, G.Y.; Griffiths, H.R. Oxidized LDL lipids increase β-amyloid production by SH-SY5Y cells through glutathione depletion and lipid raft formation. Free Radic. Biol. Med. 2014, 75, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Tamaoka, A.; Araki, W. Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J. Neurosci. Res. 2010, 88, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Dasari, B.; Prasanthi, J.R.; Marwarha, G.; Singh, B.B.; Ghribi, O. The oxysterol 27-hydroxycholesterol increases β-amyloid and oxidative stress in retinal pigment epithelial cells. BMC Ophthalmol. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Price, S.L.; Fiol-Deroque, M.A.; Marcilla-Etxenike, A.; Ahyayauch, H.; Barceló-Coblijn, G.; Terés, S.; Katsouri, L.; Ordinas, M.; López, D.J.; et al. Membrane lipid modifications and therapeutic effects mediated by hydroxydocosahexaenoic acid on Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1838, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Batten, S.E.; Harris, M.; Rockett, B.D.; Shaikh, S.R.; Stillwell, W.; Wassall, S.R. Docosahexaenoic and eicosapentaenoic acids segregate differently between raft and nonraft domains. Biophys. J. 2012, 103, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Trøseid, M.; Arnesen, H.; Hjerkinn, E.M.; Seljeflot, I. Serum levels of interleukin-18 are reduced by diet and n-3 fatty acid intervention in elderly high-risk men. Metabolism 2009, 58, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Leder, L.; Kolehmainen, M.; Narverud, I.; Dahlman, I.; Myhrstad, M.C.; de Mello, V.D.; Paananen, J.; Carlberg, C.; Schwab, U.; Herzig, K.H.; et al. Effects of a healthy Nordic diet on gene expression changes in peripheral blood mononuclear cells in response to an oral glucose tolerance test in subjects with metabolic syndrome: A SYSDIET sub-study. Genes Nutr. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, G.H.; Lee, Y.K.; Park, J.H. Changes in the arginine methylation of organ proteins during the development of diabetes mellitus. Diabetes Res. Clin. Pract. 2011, 94, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Brixius, K.; Brinkmann, C. Exercise for the diabetic brain: How physical training may help prevent dementia and Alzheimer’s disease in T2DM patients. Endocrine 2016, 53, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla, E.P.; Conti, B. Interleukin-18 null mutation increases weight and food intake and reduces energy expenditure and lipid substrate utilization in high-fat diet fed mice. Brain Behav. Immun. 2014, 3, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Hoelzer, D.; Ottmann, O.G. Regulation of human IL-18 gene expression: Interaction of PU.1 with GC-box binding protein is involved in human IL-18 expression in myeloid cells. Eur. J. Immunol. 2004, 34, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Kalina, U.; Ballas, K.; Koyama, N.; Kauschat, D.; Miething, C.; Arnemann, J.; Martin, H.; Hoelzer, D.; Ottmann, O.G. Genomic organization and regulation of the human interleukin-18 gene. Scand. J Immunol. 2000, 52, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Dwarakanath, R.S.; Reddy, M.A.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth muscle cells: A novel cross-talk in the pathogenesis of atherosclerosis. Circ. Res. 2005, 96, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.; Massamiri, T.; Deng, T.; Dewji, N.N.; Bayney, R.M.; Brown, J.H. A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer’s beta-amyloid precursor protein gene. J. Biol. Chem. 1994, 269, 21682–21690. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojala, J.O.; Sutinen, E.M. The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease. J. Clin. Med. 2017, 6, 55. https://doi.org/10.3390/jcm6050055
Ojala JO, Sutinen EM. The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease. Journal of Clinical Medicine. 2017; 6(5):55. https://doi.org/10.3390/jcm6050055
Chicago/Turabian StyleOjala, Johanna O., and Elina M. Sutinen. 2017. "The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease" Journal of Clinical Medicine 6, no. 5: 55. https://doi.org/10.3390/jcm6050055
APA StyleOjala, J. O., & Sutinen, E. M. (2017). The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease. Journal of Clinical Medicine, 6(5), 55. https://doi.org/10.3390/jcm6050055