The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis


{kind=link}
Abstract
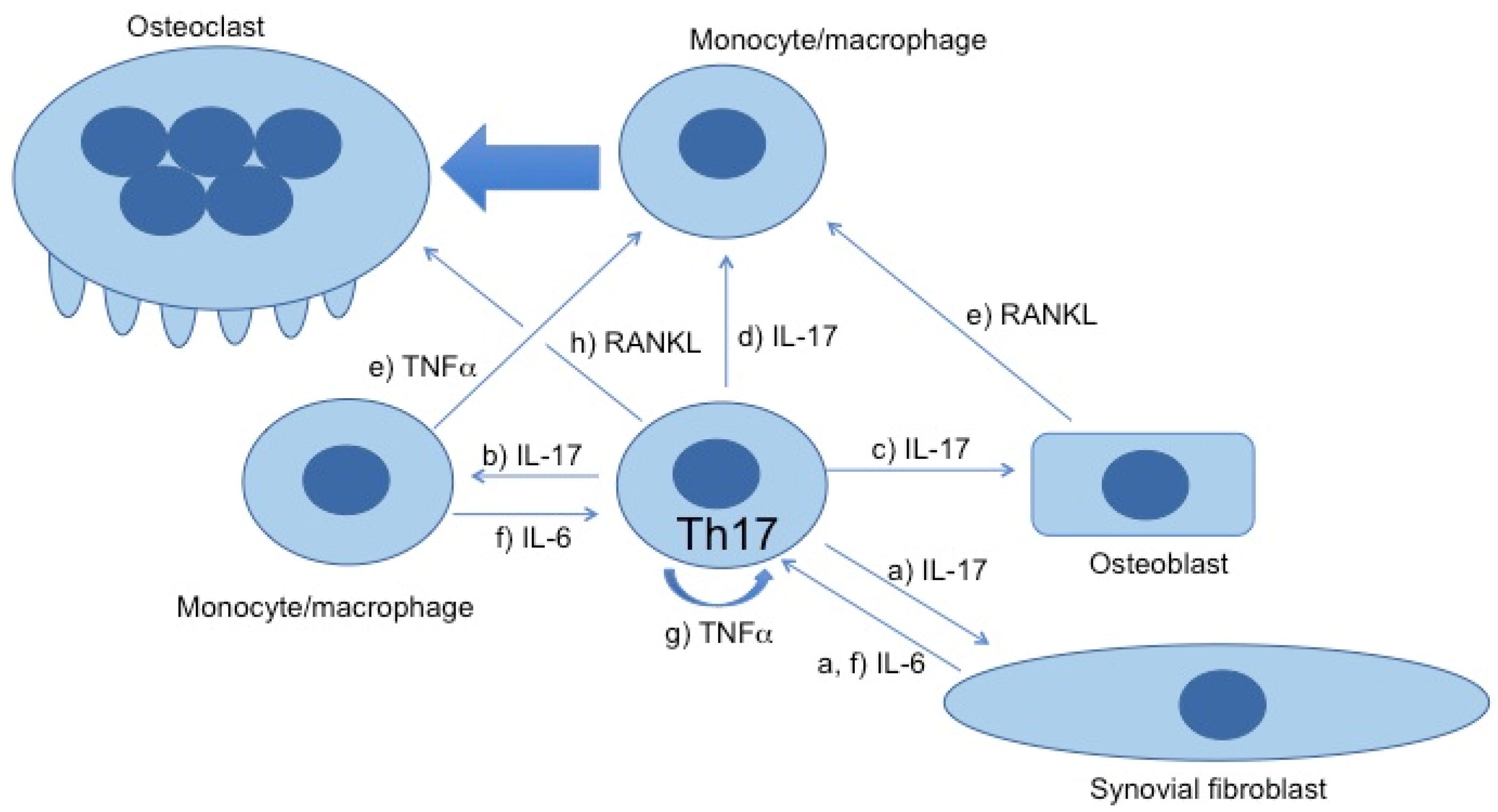
:1. Introduction
2. Plasticity of Th17
3. Th17-Derived Th1 Cells (CD161+ Th1 Cells, Nonclassic Th1 Cells)
4. Th17-Producing IFNγ (IFNγ+ Th17)
5. Possible Implications for Novel Therapies
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Kotake, S.; Nanke, Y.; Mogi, M.; Kawamoto, M.; Furuya, T.; Yago, T.; Kobashigawa, T.; Togari, A.; Kamatani, N. IFN-γ-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur. J. Immunol. 2005, 35, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar] [PubMed]
- Jovanovic, D.V.; DiBattista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.-P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-1β and TNFα, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Yago, T.; Nanke, Y.; Ichikawa, N.; Kobashigawa, T.; Mogi, M.; Kamatani, N.; Kotake, S. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: A novel mechanism of osteoclastogenesis by IL-17. J. Cell. Biochem. 2009, 108, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Audia, S.; Janikashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Cimaz, R.; Capone, M.; Santarlasci, V.; Querci, V.; Simonini, G.; Nencini, F.; Liotta, F.; Romagnani, S.; Maggi, E.; et al. Brief report: Etanercept inhibits the tumor necrosis factor α-driven shift of Th17 lymphocytes toward a nonclassic Th1 phenotype in juvenile idiopathic arthritis. Arthritis Rheumatol. 2014, 66, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, J.; Wada, Y.; Kowada, T.; Wang, Z.; Sun-Wada, G.H.; Nishiyama, I.; Mizukami, S.; Maiya, N.; Yasuda, H.; Kumanogoh, A.; et al. Dynamic visualization of RANKL and Th17-mediated osteoclast function. J. Clin. Investig. 2013, 123, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Raza, K.; Falciani, F.; Curnow, S.J.; Ross, E.J.; Lee, C.Y.; Akbar, A.N.; Lord, J.M.; Gordon, C.; Buckley, C.D.; Salmon, M. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res. Ther. 2005, 7, R784–R795. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, H.; Söderström, I.; Rocklöv, J.; Hallmans, G.; Lejon, K.; Dahlqvist, S.R. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Chalan, P.; Kroesen, B.J.; van der Geest, K.S.; Huitema, M.G.; Abdulahad, W.H.; Bijzet, J.; Brouwer, E.; Boots, A.M. Circulating CD4+CD161+ T lymphocytes are increased in seropositive arthralgia patients but decreased in patients with newly diagnosed rheumatoid arthritis. PLoS ONE 2013, 8, e79370. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Kochi, Y.; Okada, Y.; Suzuki, A.; Ikari, K.; Terao, C.; Takahashi, A.; Yamazaki, K.; Hosono, N.; Myouzen, K.; Tsunoda, T.; et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat. Genet. 2010, 42, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Binger, K.J.; Côrte-Real, B.F.; Kleinewietfeld, M. Immunometabolic Regulation of Interleukin-17-Producing T Helper Cells: Uncoupling New Targets for Autoimmunity. Front. Immunol. 2017, 8, 311. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Cimaz, R.; Maggi, L.; Santarlasci, V.; Capone, M.; Borriello, F.; Frosali, F.; Querci, V.; Simonini, G.; Barra, G.; et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S.; Annunziato, F. Th17 and nonclassic Th1 cells in chronic inflammatory disorders: Two sides of the same coin. Int. Arch. Allergy Immunol. 2014, 164, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Human Th1 dichotomy: Origin, phenotype and biologic activities. Immunology 2014, 144, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Santarlasci, V.; Capone, M.; Rossi, M.C.; Querci, V.; Mazzoni, A.; Cimaz, R.; De Palma, R.; Liotta, F.; Maggi, E.; et al. Distinctive features of classic and nonclassic (Th17 derived) human Th1 cells. Eur. J. Immunol. 2012, 42, 3180–3188. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- Firestein, G.S.; Zvaifler, N.J. Peripheral blood and synovial fluid monocyte activation in inflammatory arthritis. II. Low levels of synovial fluid and synovial tissue interferon suggest that gamma-interferon is not the primary macrophage activating factor. Arthritis Rheum. 1987, 30, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Zvaifler, N.J. How important are T cells in chronic rheumatoid synovitis. Arthritis Rheum. 1990, 33, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.A. The role of T cells in the immunopathogenesis of rheumatoid arthritis: New perspectives. Arthritis Rheum. 1997, 40, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Laurie, E.; Harrington, L.E.; Paul, R.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An Effector CD4 T Cell Lineage with Regulatory T Cell Ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Khader, S.A.; Gopal, R. IL-17 in protective immunity to intracellular pathogens. Virulence 2010, 1, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Furuya, T.; Kobashigawa, T.; Kamatani, N.; Kotake, S. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res. Ther. 2007, 9, R96. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Filì, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; De Palma, R.; Santarlasci, V.; Maggi, L.; Capone, M.; Frosali, F.; Rodolico, G.; Querci, V.; Abbate, G.; Angeli, R.; et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J. Exp. Med. 2008, 205, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Nanke, Y.; Kobashigawa, T.; Yago, T.; Kawamoto, M.; Yamanaka, H.; Kotake, S. Detection of IFN-γ+IL-17+ cells in salivary glands of patients with Sjögren’s syndrome and Mikulicz’s disease: Potential role of Th17 Th1 in the pathogenesis of autoimmune diseases. Nihon Rinsho Meneki Gakkai Kaishi 2016, 39, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, M.E.; Mendoza, L.; Villarreal, C.; Alvarez-Buylla, E.R. A minimal regulatory network of extrinsic and intrinsic factors recovers observed patterns of CD4+ T cell differentiation and plasticity. PLoS Comput. Biol. 2015, 11, e1004324. [Google Scholar] [CrossRef] [PubMed]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.P.; Cao, A.T.; Feng, T.; Li, Q.; Zhang, W.; Yao, S.; Dann, S.M.; Elson, C.O.; Cong, Y. TGF-β converts Th1 cells into Th17 cells through stimulation of Runx1 expression. Eur. J. Immunol. 2015, 45, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Geginat, J.; Paroni, M.; Kastirr, I.; Larghi, P.; Pagani, M.; Abrignani, S. Reverse plasticity: TGF-β and IL-6 induce Th1-to-Th17-cell transdifferentiation in the gut. Eur. J. Immunol. 2016, 46, 2306–2310. [Google Scholar] [CrossRef] [PubMed]
- Kleinschek, M.A.; Boniface, K.; Sadekova, S.; Grein, J.; Murphy, E.E.; Turner, S.P.; Raskin, L.; Desai, B.; Faubion, W.A.; de Waal Malefyt, R.; et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 2009, 206, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.B.; Bettadapura, J.; Alsharifi, M.; Mathew, P.A.; Warren, H.S.; Lanier, L.L. Lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J. Immunol. 2005, 175, 7796–7799. [Google Scholar] [CrossRef] [PubMed]
- Huarte, E.; Cubillos-Ruiz, J.R.; Nesbeth, Y.C.; Scarlett, U.K.; Martinez, D.G.; Engle, X.A.; Rigby, W.F.; Pioli, P.A.; Guyre, P.M.; Conejo-Garcia, J.R. PILAR is a novel modulator of human T-cell expansion. Blood 2008, 112, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Capone, M.; Giudici, F.; Santarlasci, V.; Querci, V.; Liotta, F.; Ficari, F.; Maggi, E.; Tonelli, F.; Annunziato, F.; et al. CD4+CD161+ T lymphocytes infiltrate Crohn’s disease-associated perianal fistulas and are reduced by anti-TNF-α local therapy. Int. Arch. Allergy Immunol. 2013, 161, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Margheri, F.; Luciani, C.; Capone, M.; Rossi, M.C.; Chillà, A.; Santarlasci, V.; Mazzoni, A.; Cimaz, R.; Liotta, F.; et al. Th1-Induced CD106 expression mediates leukocytes adhesion on synovial fibroblasts from juvenile idiopathic arthritis patients. PLoS ONE 2016, 11, e0154422. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Martin-Orozco, N.; Chung, Y.; Chang, S.H.; Wang, Y.H.; Dong, C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur. J. Immunol. 2009, 39, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Bending, D.; De la Peña, H.; Veldhoen, M.; Phillips, J.M.; Uyttenhove, C.; Stockinger, B.; Cooke, A. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J. Clin. Investig. 2009, 119, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Zhang, K.; Lv, M.; LI, Q.; Zheng, Z.; Han, Q.; Guo, N.; Fan, C.; Zhu, P. Circulating Th17 and Th1 cells expressing CD161 are associated with disease activity in rheumatoid arthritis. Scand. J. Rheumatol. 2014, 43, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Nakashima, Y.; Okazaki, K.; Mawatari, T.; Fukushi, J.I.; Kaibara, N.; Hori, A.; Iwamoto, Y.; Yoshikai, Y. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Nanke, Y.; Yago, T.; Kawamoto, M.; Kobashigawa, T.; Yamanaka, H. Elevated Ratio of Th17 Cell-Derived Th1 Cells (CD161(+)Th1 Cells) to CD161(+)Th17 Cells in Peripheral Blood of Early-Onset Rheumatoid Arthritis Patients. BioMed Res. Int. 2016, 2016, 4186027. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Nanke, Y.; Yago, T.; Kawamoto, M.; Kobashigawa, T.; Yamanaka, H. Ratio of circulating IFNγ (+) “Th17 Cells” in memory Th cells is inversely correlated with the titer of anti-CCP antibodies in early-onset rheumatoid arthritis patients based on flow cytometry methods of the human immunology project. BioMed Res. Int. 2016, 2016, 9694289. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Mazurov, V.; Aelion, J.A.; Lee, S.H.; Codding, C.E.; Kellner, H.; et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: A phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann. Rheum. Dis. 2013, 72, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Greenwald, M.; Cho, C.S.; Berman, A.; Jin, L.; Cameron, G.S.; Benichou, O.; Xie, L.; Braun, D.; Berclaz, P.Y.; et al. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014, 66, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; Patel, D.D.; Dryja, T.; Wright, A.M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M.H.; Psoriasis Study Group; et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2010, 2, 52ra72. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Durez, P.; Shestakova, G.; Genovese, M.C.; Schulze-Koops, H.; Li, Y.; Wang, Y.A.; Lewitzky, S.; Koroleva, I.; Berneis, A.A.; et al. Association of HLA-DRB1 alleles with clinical responses to the anti-interleukin-17A monoclonal antibody secukinumab in active rheumatoid arthritis. Rheumatology 2016, 55, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Aelion, J.A.; Lee, S.H.; Codding, C.E.; Kellner, H.; Ikawa, T.; et al. One-year efficacy and safety results of secukinumab in patients with rheumatoid arthritis: Phase II, dose-finding, double-blind, randomized, placebo-controlled study. J. Rheumatol. 2014, 41, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Braun, D.K.; Erickson, J.S.; Berclaz, P.Y.; Banerjee, S.; Heffernan, M.P.; Carlier, H. Safety and Efficacy of Open-label Subcutaneous Ixekizumab Treatment for 48 Weeks in a Phase II Study in Biologic-naive and TNF-IR Patients with Rheumatoid Arthritis. J. Rheumatol. 2016, 43, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Möricke, R.; Dokoupilova, E.; Codding, C.; Neal, J.; Andersson, M.; Rohrer, S.; Richards, H. Secukinumab in active rheumatoid arthritis: A randomized, double-blind placebo and active comparator controlled phase 3 study. Arthritis Rheumatol. 2017, 69, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Duan, D. Efficacy and safety of monoclonal antibodies targeting interleukin-17 pathway for inflammatory arthritis: A meta-analysis of randomized controlled clinical trials. Drug Des. Dev. Ther. 2016, 10, 2771–2777. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Dahal, K.; Sharma, S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: A systematic literature review and meta-analysis of randomized controlled trials. Rheumatol. Int. 2016, 36, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.C. Associations between circulating IL-17 levels and rheumatoid arthritis and between IL-17 gene polymorphisms and disease susceptibility: A meta-analysis. Postgrad. Med. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.A.; Hueber, A.J.; Wilson, S.; Galm, M.; Baum, W.; Kitson, C.; Auer, J.; Lorenz, S.H.; Moelleken, J.; Bader, M.; et al. Combined inhibition of tumor necrosis factor α and interleukin-17 as a therapeutic opportunity in rheumatoid arthritis: Development and characterization of a novel bispecific antibody. Arthritis Rheumatol. 2015, 67, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Buckland, J. Rheumatoid arthritis: Anti-TNF and anti-IL-17 antibodies—Better together! Nat. Rev. Rheumatol. 2014, 10, 699. [Google Scholar] [CrossRef] [PubMed]
- Pauthner, M.; Yeung, J.; Ullman, C.; Bakker, J.; Wurch, T.; Reichert, J.M.; Lund-Johansen, F.; Bradbury, A.R.; Carter, P.J.; Melis, J.P. Antibody engineering & therapeutics, the annual meeting of the antibody society December 7–10, 2015, San Diego, CA, USA. MAbs 2016, 8, 617–652. [Google Scholar] [PubMed]
- Silacci, M.; Lembke, W.; Woods, R.; Attinger-Toller, I.; Baenziger-Tobler, N.; Batey, S.; Santimaria, R.; von der Bey, U.; Koenig-Friedrich, S.; Zha, W.; et al. Discovery and characterization of COVA322, a clinical-stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. MAbs 2016, 8, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ndongo-Thiam, N.; Miossec, P. A cell-based bioassay for circulating bioactive IL-17: Application to destruction in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1629–1631. [Google Scholar] [CrossRef] [PubMed]
- Merola, J.F.; Lockshin, B.; Mody, E.A. Switching biologics in the treatment of psoriatic arthritis. Semin. Arthritis Rheum. 2017. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotake, S.; Yago, T.; Kobashigawa, T.; Nanke, Y. The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 67. https://doi.org/10.3390/jcm6070067
Kotake S, Yago T, Kobashigawa T, Nanke Y. The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. Journal of Clinical Medicine. 2017; 6(7):67. https://doi.org/10.3390/jcm6070067
Chicago/Turabian StyleKotake, Shigeru, Toru Yago, Tsuyoshi Kobashigawa, and Yuki Nanke. 2017. "The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis" Journal of Clinical Medicine 6, no. 7: 67. https://doi.org/10.3390/jcm6070067
APA StyleKotake, S., Yago, T., Kobashigawa, T., & Nanke, Y. (2017). The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. Journal of Clinical Medicine, 6(7), 67. https://doi.org/10.3390/jcm6070067