Lipid Myopathies


Abstract
:1. Introduction
2. Diseases
2.1. Primary Carnitine Deficiency (PCD)
2.1.1. Clinical Presentation
2.1.2. Laboratory Data and Diagnosis
2.1.3. Therapy
2.2. Neutral Lipid Storage Disease (NLSD)
2.2.1. Clinical Presentation
2.2.2. Laboratory Data and Diagnosis
2.2.3. Therapy
2.3. Phosphatidic Acid Phosphatase Deficiency (Lipin Deficiency)
2.3.1. Clinical Presentation
2.3.2. Laboratory Data and Diagnosis
2.3.3. Therapy
2.4. Carnitine-Acylcarnitine Translocase (CACT) Deficiency
2.4.1. Clinical Presentation
2.4.2. Laboratory Data and Diagnosis
2.4.3. Therapy
2.5. Carnitine Palmitoyl Transferase II (CPT II) Deficiency
2.5.1. Clinical Presentation
2.5.2. Laboratory Data and Diagnosis
2.5.3. Therapy
2.6. Very-Long-Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency
2.6.1. Clinical Presentation
2.6.2. Laboratory Data and Diagnosis
2.6.3. Therapy
2.7. Acyl-CoA Dehydrogenase 9 (ACAD9) Deficiency
2.7.1. Clinical Presentation
2.7.2. Laboratory Data and Diagnosis
2.7.3. Therapy
2.8. Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency
2.8.1. Clinical Presentation
2.8.2. Laboratory Data and Diagnosis
2.8.3. Therapy
2.9. Short-Chain Acyl-CoA Dehydrogenase (SCAD) Deficiency
2.9.1. Clinical Presentation
2.9.2. Laboratory Data and Diagnosis
2.9.3. Therapy
2.10. Mitochondrial Trifunctional Protein (MTP) Deficiency
2.10.1. Clinical Presentation
2.10.2. Laboratory Data and Diagnosis
2.10.3. Therapy
2.11. Short-Chain L-3-Hydroxyacyl-CoA Dehydrogenase (SCHAD) Deficiency
2.11.1. Clinical Presentation
2.11.2. Laboratory Data and Diagnosis
2.11.3. Therapy
2.12. Medium-Chain 3-Ketoacyl-CoA Thiolase (MCKAT)
2.12.1. Clinical Presentation
2.12.2. Laboratory Data
2.12.3. Therapy
2.13. Multiple Acyl-CoA Dehydrogenase Deficiency (MADD)
2.13.1. Clinical Presentation
2.13.2. Laboratory Data and Diagnosis
2.13.3. Therapy
3. Acquired Disorders of Lipid Metabolism in Muscle
3.1. HIV
3.2. Alcohol
3.3. Drugs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Buhman, K.K.; Chen, H.C.; Farese, R.V. The enzymes of neutral lipid synthesis. J. Biol. Chem. 2001, 276, 40369–40372. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Nishino, I. Lipid storage myopathy. Curr. Neurol. Neurosci. Rep. 2011, 11, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Toscano, A.; Barca, E.; Musumeci, O. Update on diagnostics of metabolic myopathies. Curr. Opin. Neurol. 2017, 30, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Laforêt, P.; Vianey-Saban, C. Disorders of muscle lipid metabolism: Diagnostic and therapeutic challenges. Neuromuscul. Disord. 2010, 20, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E.M.; Missaglia, S.; Dimauro, S.; Bernardi, C.; Akman, H.O.; Tavian, D. A myopathy with unusual features caused by PNPLA2 gene mutations. Muscle Nerve 2015, 51, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Laforêt, P.; Stojkovic, T.; Bassez, G.; Carlier, P.G.; Clément, K.; Wahbi, K.; Petit, F.M.; Eymard, B.; Carlier, R.-Y. Neutral lipid storage disease with myopathy: A whole-body nuclear MRI and metabolic study. Mol. Genet. MeTable 2013, 108, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, M.; Tasca, G.; Diaz-Manera, J.; Ottaviani, P.; Laschena, F.; Pantoli, D.; Gerevini, S.; Fiorillo, C.; Maggi, L.; Tasca, E.; et al. Muscle MRI in neutral lipid storage disease (NLSD). J. Neurol. 2017, 264, 1334–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Christopher, R.; Gayathri, N.; Natarajan, A.; Manjunath, M.; Nashi, S.; Prasad, C.; Nalini, A. Fatty acid oxidation defects presenting as primary myopathy and prominent dropped head syndrome. Neuromuscul. Disord. 2017, 27, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Diekman, E.F.; van der Pol, W.L.; Nievelstein, R.A.J.; Houten, S.M.; Wijburg, F.A.; Visser, G. Muscle MRI in patients with long-chain fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2014, 37, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Bruno, C.; Dimauro, S. Lipid storage myopathies. Curr. Opin. Neurol. 2008, 21, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; De Vivo, D.C.; Bierman, F.; Pulver, P.; De Meirleir, L.J.; Cvitanovic-Sojat, L.; Pagon, R.A.; Bertini, E.; Dionisi-Vici, C.; Servidei, S. Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr. Res. 1990, 28, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Angelini, C. Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: A new syndrome. Science 1973, 179, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 1999, 21, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, A.; Nozaki, J.; Ohura, T.; Kayo, T.; Wada, Y.; Nezu, J.; Ohashi, R.; Tamai, I.; Shoji, Y.; Takada, G.; et al. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum. Mol. Genet. 1999, 8, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Novel OCTN2 Mutations: No Genotype-Phenotype Correlations: Early Carnitine Therapy Prevents Cardiomyopathy. Available online: https://www.ncbi.nlm.nih.gov/pubmed/?term=Novel+OCTN2+mutations%3A+no+genotype-phenotype+correlations%3A+early+carnitine+therapy+prevents+cardiomyopathy (accessed on 27 August 2018).
- Ohkuma, A.; Noguchi, S.; Sugie, H.; Malicdan, M.C.V.; Fukuda, T.; Shimazu, K.; López, L.C.; Hirano, M.; Hayashi, Y.K.; Nonaka, I.; et al. Clinical and genetic analysis of lipid storage myopathies. Muscle Nerve 2009, 39, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Tein, I. Lipid storage myopathies due to fatty acid oxidation defects. Neuromuscular Disorders of Infancy, Childhood, and Adolescence; Elsevier: New York, NY, USA, 2015; pp. 761–795. [Google Scholar]
- Lamhonwah, A.-M.; Olpin, S.E.; Pollitt, R.J.; Vianey-Saban, C.; Divry, P.; Guffon, N.; Besley, G.T.N.; Onizuka, R.; De Meirleir, L.J.; Cvitanovic-Sojat, L.; et al. Novel OCTN2 mutations: No genotype-phenotype correlations: Early carnitine therapy prevents cardiomyopathy. Am. J. Med. Genet. 2002, 111, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Lefèvre, C.; Morava, E.; Mussini, J.-M.; Laforêt, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, C.; Jobard, F.; Caux, F.; Bouadjar, B.; Karaduman, A.; Heilig, R.; Lakhdar, H.; Wollenberg, A.; Verret, J.L.; Weissenbach, J.; et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 2001, 69, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.; Schweiger, M.; Kloss-Brandstätter, A.; Lamina, C.; Haun, M.; Erhart, G.; Paulweber, B.; Rahman, Y.; Olpin, S.; Wolinski, H.; et al. Investigation and functional characterization of rare genetic variants in the adipose triglyceride lipase in a large healthy working population. PLoS Genet. 2010, 6, e1001239. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Ikeda, Y.; Zaima, N.; Sakata, Y.; Matsumiya, G. Triglyceride deposit cardiomyovasculopathy. N. Engl. J. Med. 2008, 359, 2396–2398. [Google Scholar] [CrossRef] [PubMed]
- Higashi, M.; Hirano, K.; Kobayashi, K.; Ikeda, Y.; Issiki, A.; Otsuka, T.; Suzuki, A.; Yamaguchi, S.; Zaima, N.; Hamada, S.; et al. Distinct cardiac phenotype between two homozygotes born in a village with accumulation of a genetic deficiency of adipose triglyceride lipase. Int. J. Cardiol. 2015, 192, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E.M.; Arca, M.; Bertini, E.; Bruno, C.; Cassandrini, D.; D’amico, A.; Garibaldi, M.; Gragnani, F.; Maggi, L.; Massa, R.; et al. Italian NLSD group neutral lipid storage diseases: Clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J. Rare. Dis. 2017, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Jordans, G.H. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva (ERB.). Acta Med. Scand. 1953, 145, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Snyder, T.M.; Little, B.W.; Roman-Campos, G.; McQuillen, J.B. Successful treatment of familial idiopathic lipid storage myopathy with L-carnitine and modified lipid diet. Neurology 1982, 32, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Weijer, T.; Havekes, B.; Bilet, L.; Hoeks, J.; Sparks, L.; Bosma, M.; Paglialunga, S.; Jorgensen, J.; Janssen, M.C.H.; Schaart, G.; et al. Effects of bezafibrate treatment in a patient and a carrier with mutations in the PNPLA2 gene, causing neutral lipid storage disease with myopathy. Circ. Res. 2013, 112, e51–e54. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A.; Roy, S.; Sarkadi-Nagy, E.; Kim, K.-H.; Sul, H.S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: Ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 2004, 279, 47066–47075. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Yang, H.; Wang, S.P.; Soni, K.G.; Brunel-Guitton, C.; Mitchell, G.A. Inborn errors of cytoplasmic triglyceride metabolism. J. Inherit. Metab. Dis. 2015, 38, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Michot, C.; Mamoune, A.; Vamecq, J.; Viou, M.T.; Hsieh, L.-S.; Testet, E.; Lainé, J.; Hubert, L.; Dessein, A.-F.; Fontaine, M.; et al. Combination of lipid metabolism alterations and their sensitivity to inflammatory cytokines in human lipin-1-deficient myoblasts. Biochim. Biophys. Acta 2013, 1832, 2103–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, G.G.; Collier, S.L.; Chen, Z.; Eaton, J.M.; Connolly, A.M.; Bucelli, R.C.; Pestronk, A.; Harris, T.E.; Finck, B.N. Rhabdomyolysis-Associated mutations in human LPIN1 lead to loss of phosphatidic acid phosphohydrolase activity. JIMD Rep. 2015, 23, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bergounioux, J.; Brassier, A.; Rambaud, C.; Bustarret, O.; Michot, C.; Hubert, L.; Arnoux, J.B.; Laquerriere, A.; Bekri, S.; Galene-Gromez, S.; et al. Fatal rhabdomyolysis in 2 children with LPIN1 mutations. J. Pediatr. 2012, 160, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Pichler, K.; Scholl-Buergi, S.; Birnbacher, R.; Freilinger, M.; Straub, S.; Brunner, J.; Zschocke, J.; Bittner, R.E.; Karall, D. A novel therapeutic approach for LPIN1 mutation-associated rhabdomyolysis—The Austrian experience. Muscle Nerve 2015, 52, 437–439. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; DiMauro, P.M. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 1973, 182, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C. Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wieser, T.; Deschauer, M.; Olek, K.; Hermann, T.; Zierz, S. Carnitine palmitoyltransferase II deficiency: Molecular and biochemical analysis of 32 patients. Neurology 2003, 60, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Carnitine palmitoyltransferase II (CPT II) deficiency: Genotype-phenotype analysis of 50 patients. J. Neurol. Sci. 2014, 338, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Brucknerova, I.; Bzduch, V.; Behulova, D.; Ferianec, V.; Dubovicky, M.; Ujhazy, E.; Mach, M. Reversible asphyxial status in a newborn due to neonatal form of carnitine palmitoyltransferase II deficiency. Neuro Endocrinol. Lett. 2008, 29, 627–630. [Google Scholar] [PubMed]
- Tajima, G.; Hara, K.; Tsumura, M.; Kagawa, R.; Okada, S.; Sakura, N.; Maruyama, S.; Noguchi, A.; Awaya, T.; Ishige, M.; et al. Newborn screening for carnitine palmitoyltransferase II deficiency using (C16+C18:1)/C2: Evaluation of additional indices for adequate sensitivity and lower false-positivity. Mol. Genet. MeTable 2017, 122, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.R.; Deschauer, M.; Zierz, S. Clinically symptomatic heterozygous carnitine palmitoyltransferase II (CPT II) deficiency. Wien. Klin. Wochenschr. 2012, 124, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Ørngreen, M.C.; Ejstrup, R.; Vissing, J. Effect of diet on exercise tolerance in carnitine palmitoyltransferase II deficiency. Neurology 2003, 61, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Ørngreen, M.C.; Madsen, K.L.; Preisler, N.; Andersen, G.; Vissing, J.; Laforêt, P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: A randomized clinical trial. Neurology 2014, 82, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Vianey-Saban, C.; Bross, P.; Divry, P.; Roe, C.R.; Nada, M.A.; Knudsen, I.; Gregersen, N. The mutational spectrum in very long-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 1996, 19, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Indo, Y.; Coates, P.M.; Hashimoto, T.; Tanaka, K. Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Pediatr. Res. 1993, 34, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Tein, I. Disorders of fatty acid oxidation. In Handbook of Clinical Neurology; Elsevier: New York, NY, USA, 2013; Volume 113, pp. 1165–1688. [Google Scholar]
- Laforêt, P.; Acquaviva-Bourdain, C.; Rigal, O.; Brivet, M.; Penisson-Besnier, I.; Chabrol, B.; Chaigne, D.; Boespflug-Tanguy, O.; Laroche, C.; Bedat-Millet, A.-L.; et al. Diagnostic assessment and long-term follow-up of 13 patients with Very Long-Chain Acyl-Coenzyme A dehydrogenase (VLCAD) deficiency. Neuromuscul. Disord. 2009, 19, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Vianey-Saban, C.; Divry, P.; Brivet, M.; Nada, M.; Zabot, M.T.; Mathieu, M.; Roe, C. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: Clinical characteristics and diagnostic considerations in 30 patients. Clin. Chim. Acta 1998, 269, 43–62. [Google Scholar] [CrossRef]
- Merinero, B.; Alcaide, P.; Martín-Hernández, E.; Morais, A.; García-Silva, M.T.; Quijada-Fraile, P.; Pedrón-Giner, C.; Dulin, E.; Yahyaoui, R.; Egea, J.M.; et al. Four years’ experience in the diagnosis of very long-chain Acyl-CoA dehydrogenase deficiency in infants detected in three Spanish newborn screening centers. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2017; Volume 39, pp. 63–74. [Google Scholar]
- Roe, C.R.; Sweetman, L.; Roe, D.S.; David, F.; Brunengraber, H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Investig. 2002, 110, 259–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, A.M.; Conlon, T.; Walter, G.; Zeng, H.; Shaffer, S.A.; Dungtao, F.; Erger, K.; Cossette, T.; Tang, Q.; Mueller, C.; et al. Long-term correction of very long-chain Acyl-CoA dehydrogenase deficiency in mice using AAV9 gene therapy. Mol. Ther. 2012, 20, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Swigonová, Z.; Mohsen, A.-W.; Vockley, J. Acyl-CoA dehydrogenases: Dynamic history of protein family evolution. J. Mol. Evol. 2009, 69, 176–193. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Rutledge, S.L.; Kelly, D.R.; Palmer, C.A.; Murdoch, G.; Majumder, N.; Nicholls, R.D.; Pei, Z.; Watkins, P.A.; Vockley, J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am. J. Hum. Genet. 2007, 81, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Gerards, M.; van den Bosch, B.J.C.; Danhauser, K.; Serre, V.; van Weeghel, M.; Wanders, R.J.A.; Nicolaes, G.A.F.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: New function for an old gene. Brain 2011, 134, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Lund, A.M.; Hougaard, D.M.; Christensen, E.; Gahrn, B.; Christensen, M.; Bross, P.; Vested, A.; Simonsen, H.; Skogstrand, K.; et al. MCAD deficiency in Denmark. Mol. Genet. MeTable 2012, 106, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Ziadeh, R.; Hoffman, E.P.; Finegold, D.N.; Hoop, R.C.; Brackett, J.C.; Strauss, A.W.; Naylor, E.W. Medium chain Acyl-CoA dehydrogenase deficiency in Pennsylvania: Neonatal screening shows high incidence and unexpected mutation frequencies. Pediatr. Res. 1995, 37, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Rhead, W.J. Newborn screening for medium-chain Acyl-CoA dehydrogenase deficiency: A global perspective. J. Inherit. Metab. Dis. 2006, 29, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Tajima, G.; Hara, K.; Tsumura, M.; Kagawa, R.; Okada, S.; Sakura, N.; Hata, I.; Shigematsu, Y.; Kobayashi, M. Screening of MCAD deficiency in Japan: 16 Years’ experience of enzymatic and genetic evaluation. Mol. Genet. Metab. 2016, 119, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Mayell, S.J.; Edwards, L.; Reynolds, F.E.; Chakrapani, A.B. Late presentation of medium-chain Acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2007, 30, 104. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A.; Hale, D.E.; Coates, P.M. Medium-chain Acyl-CoA dehydrogenase deficiency. Prog. Clin. Biol. Res. 1990, 321, 291–302. [Google Scholar] [PubMed]
- Nelson, J.; Lewis, B.; Walters, B. The HELLP syndrome associated wiht fetal medium-chain Acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2000, 23, 518–519. [Google Scholar] [CrossRef] [PubMed]
- Innes, A.M.; Seargeant, L.E.; Balachandra, K.; Roe, C.R.; Wanders, R.J.; Ruiter, J.P.; Casiro, O.; Grewar, D.A.; Greenberg, C.R. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr. Res. 2000, 47, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Matern, D.; Hart, P.; Murtha, A.P.; Vockley, J.; Gregersen, N.; Millington, D.S.; Treem, W.R. Acute fatty liver of pregnancy associated with short-chain acyl-coenzyme A dehydrogenase deficiency. J. Pediatr. 2001, 138, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Treem, W.R.; Stanley, C.A.; Goodman, S.I. Medium-chain acyl-CoA dehydrogenase deficiency: Metabolic effects and therapeutic efficacy of long-term L-carnitine supplementation. J. Inherit. Metab. Dis. 1989, 12, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.R.; Millington, D.S.; Maltby, D.A.; Kinnebrew, P. Recognition of medium-chain Acyl-CoA dehydrogenase deficiency in asymptomatic siblings of children dying of sudden infant death or Reye-like syndromes. J. Pediatr. 1986, 108, 13–18. [Google Scholar] [CrossRef]
- Bhala, A.; Willi, S.M.; Rinaldo, P.; Bennett, M.J.; Schmidt-Sommerfeld, E.; Hale, D.E. Clinical and biochemical characterization of short-chain acyl-coenzyme A dehydrogenase deficiency. J. Pediatr. 1995, 126, 910–915. [Google Scholar] [CrossRef]
- Tein, I.; Haslam, R.H.; Rhead, W.J.; Bennett, M.J.; Becker, L.E.; Vockley, J. Short-chain Acyl-CoA dehydrogenase deficiency: A cause of ophthalmoplegia and multicore myopathy. Neurology 1999, 52, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Ribes, A.; Riudor, E.; Garavaglia, B.; Martinez, G.; Arranz, A.; Invernizzi, F.; Briones, P.; Lamantea, E.; Sentís, M.; Barceló, A.; et al. Mild or absent clinical signs in twin sisters with short-chain Acyl-CoA dehydrogenase deficiency. Eur. J. Pediatr. 1998, 157, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.B.; Kølvraa, S.; Kølvraa, A.; Stenbroen, V.; Kjeldsen, M.; Ensenauer, R.; Tein, I.; Matern, D.; Rinaldo, P.; Vianey-Saban, C.; et al. The ACADS gene variation spectrum in 114 patients with short-chain Acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum. Genet. 2008, 124, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Winter, V.S.; Corydon, M.J.; Corydon, T.J.; Rinaldo, P.; Ribes, A.; Martinez, G.; Bennett, M.J.; Vianey-Saban, C.; Bhala, A.; et al. Identification of four new mutations in the short-chain Acyl-CoA dehydrogenase (SCAD) gene in two patients: One of the variant alleles, 511C-->T, is present at an unexpectedly high frequency in the general population, as was the case for 625G-->A, together conferring susceptibility to ethylmalonic aciduria. Hum. Mol. Genet. 1998, 7, 619–627. [Google Scholar] [PubMed]
- Liu, X.; Wu, L.; Deng, G.; Li, N.; Chu, X.; Guo, F.; Li, D. Characterization of mitochondrial trifunctional protein and its inactivation study for medicine development. Biochim. Biophys. Acta 2008, 1784, 1742–1749. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Vreken, P.; den Boer, M.E.; Wijburg, F.A.; van Gennip, A.H.; IJlst, L. Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J. Inherit. Metab. Dis. 1999, 22, 442–487. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T. Peroxisomal and mitochondrial enzymes. Prog. Clin. Biol. Res. 1992, 375, 19–32. [Google Scholar] [PubMed]
- Kamijo, T.; Wanders, R.J.; Saudubray, J.M.; Aoyama, T.; Komiyama, A.; Hashimoto, T. Mitochondrial trifunctional protein deficiency. Catalytic heterogeneity of the mutant enzyme in two patients. J. Clin. Investig. 1994, 93, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Kler, R.S.; Bartlett, K.; Briggs, H.; Bindoff, L.A.; Pourfarzam, M.; Gardner-Medwin, D.; Turnbull, D.M. Combined enzyme defect of mitochondrial fatty acid oxidation. J. Clin. Invest. 1992, 90, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef] [PubMed]
- Olpin, S.E.; Clark, S.; Andresen, B.S.; Bischoff, C.; Olsen, R.K.J.; Gregersen, N.; Chakrapani, A.; Downing, M.; Manning, N.J.; Sharrard, M.; et al. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J. Inherit. Metab. Dis. 2005, 28, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Duran, M.; Wanders, R.J.; de Jager, J.P.; Dorland, L.; Bruinvis, L.; Ketting, D.; Ijlst, L.; van Sprang, F.J. 3-Hydroxydicarboxylic aciduria due to long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency associated with sudden neonatal death: Protective effect of medium-chain triglyceride treatment. Eur. J. Pediatr. 1991, 150, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Leung, K.C.; Hammond, J.; Kamath, R.; Leonard, J.V. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet 1993, 341, 407–408. [Google Scholar] [CrossRef]
- Treem, W.R.; Shoup, M.E.; Hale, D.E.; Bennett, M.J.; Rinaldo, P.; Millington, D.S.; Stanley, C.A.; Riely, C.A.; Hyams, J.S. Acute fatty liver of pregnancy, hemolysis, elevated liver enzymes, and low platelets syndrome, and long chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am. J. Gastroenterol. 1996, 91, 2293–2300. [Google Scholar] [PubMed]
- Rocchiccioli, F.; Wanders, R.J.; Aubourg, P.; Vianey-Liaud, C.; Ijlst, L.; Fabre, M.; Cartier, N.; Bougneres, P.F. Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: A cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr. Res. 1990, 28, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Bennett, M.J.; Ben-Zeev, B.; Strauss, A.W.; Tein, I. Peripheral neuropathy, episodic myoglobinuria, and respiratory failure in deficiency of the mitochondrial trifunctional protein. Muscle Nerve 2004, 29, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)? Pediatr. Neurol. 1995, 12, 21–30. [Google Scholar] [CrossRef]
- Das, A.M.; Fingerhut, R.; Wanders, R.J.; Ullrich, K. Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Possible diagnostic pitfalls. Eur. J. Pediatr. 2000, 159, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Majander, A.; Kalimo, H.; Rapola, J.; Pihko, H. Pathology of skeletal muscle and impaired respiratory chain function in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency with the G1528C mutation. Neuromuscul. Disord. 1996, 6, 327–337. [Google Scholar] [CrossRef]
- Harding, C.O.; Gillingham, M.B.; van Calcar, S.C.; Wolff, J.A.; Verhoeve, J.N.; Mills, M.D. Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U.; Lindner, M.; Santer, R.; Grotzke, M.; Baumgartner, M.R.; Boehles, H.; Das, A.; Haase, C.; Hennermann, J.B.; Karall, D.; et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: Results from a workshop. J. Inherit. Metab. Dis. 2009, 32, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Djouadi, F.; Bastin, J. PPARs as therapeutic targets for correction of inborn mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2008, 31, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Vredendaal, P.J.; van den Berg, I.E.; Malingré, H.E.; Stroobants, A.K.; Olde Weghuis, D.E.; Berger, R. Human short-chain L-3-hydroxyacyl-CoA dehydrogenase: Cloning and characterization of the coding sequence. Biochem. Biophys. Res. Commun. 1996, 223, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y.; He, X.-Y.; Schulz, H. 3-Hydroxyacyl-CoA dehydrogenase and short chain 3-hydroxyacyl-CoA dehydrogenase in human health and disease. FEBS J. 2005, 272, 4874–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tein, I.; De Vivo, D.C.; Hale, D.E.; Clarke, J.T.; Zinman, H.; Laxer, R.; Shore, A.; DiMauro, S. Short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency in muscle: A new cause for recurrent myoglobinuria and encephalopathy. Ann. Neurol. 1991, 30, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Weinberger, M.J.; Kobori, J.A.; Rinaldo, P.; Burlina, A.B. Mitochondrial short-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: A new defect of fatty acid oxidation. Pediatr. Res. 1996, 39, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J.; Spotswood, S.D.; Ross, K.F.; Comfort, S.; Koonce, R.; Boriack, R.L.; IJlst, L.; Wanders, R.J. Fatal hepatic short-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: Clinical, biochemical, and pathological studies on three subjects with this recently identified disorder of mitochondrial beta-oxidation. Pediatr. Dev. Pathol. 1999, 2, 337–345. [Google Scholar] [PubMed]
- Molven, A.; Matre, G.E.; Duran, M.; Wanders, R.J.; Rishaug, U.; Njølstad, P.R.; Jellum, E.; Søvik, O. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 2004, 53, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Indo, Y.; Souri, M.; Aoyama, T.; Hara, T.; Yamamoto, S.; Ushikubo, S.; Rinaldo, P.; Matsuda, I.; Komiyama, A.; et al. Medium chain 3-ketoacyl-coenzyme A thiolase deficiency: A new disorder of mitochondrial fatty acid beta-oxidation. Pediatr. Res. 1997, 42, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.I.; Bemelen, K.F.; Frerman, F.E. Human cDNA encoding ETF dehydrogenase (ETF: Ubiquinone oxido-reductase), and mutations in glutaric acidemia type II. Prog. Clin. Biol. Res. 1992, 375, 567–572. [Google Scholar] [PubMed]
- Wilson, G.N.; de Chadarévian, J.P.; Kaplan, P.; Loehr, J.P.; Frerman, F.E.; Goodman, S.I. Glutaric aciduria type II: Review of the phenotype and report of an unusual glomerulopathy. Am. J. Med. Genet. 1989, 32, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Grünert, S.C. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Béhin, A.; Acquaviva-Bourdain, C.; Souvannanorath, S.; Streichenberger, N.; Attarian, S.; Bassez, G.; Brivet, M.; Fouilhoux, A.; Labarre-Villa, A.; Laquerrière, A.; et al. Multiple acyl-CoA dehydrogenase deficiency (MADD) as a cause of late-onset treatable metabolic disease. Rev. Neurol. 2016, 172, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Angle, B.; Burton, B.K. Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol. Genet. MeTable 2008, 93, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Singla, M.; Guzman, G.; Griffin, A.J.; Bharati, S. Cardiomyopathy in multiple Acyl-CoA dehydrogenase deficiency: A clinico-pathological correlation and review of literature. Pediatr. Cardiol. 2008, 29, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.H.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, R.K.J.; Andresen, B.S.; Christensen, E.; Bross, P.; Skovby, F.; Gregersen, N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2003, 22, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.J.; Rodrigues, J.V.; Olsen, R.K.; Bross, P.; Gomes, C.M. Role of flavinylation in a mild variant of multiple acyl-CoA dehydrogenation deficiency: A molecular rationale for the effects of riboflavin supplementation. J. Biol. Chem. 2009, 284, 4222–4229. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.J.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.S.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, G.; Yonezawa, A.; Masuda, S.; Inui, K.; Sim, K.G.; Carpenter, K.; Olsen, R.K.J.; Mitchell, J.J.; Rhead, W.J.; Peters, G.; et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat. 2011, 32, E1976–E1984. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Abeling, N.G.G.M.; IJlst, L.; Knoester, H.; van der Pol, W.L.; Stroomer, A.E.M.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2011, 34, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Makowski, C.; Yao, Y.; Graf, E.; Hempel, M.; Wieland, T.; Tauer, U.; Ahting, U.; Mayr, J.A.; Freisinger, P.; et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012, 35, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SLC25A32 Mutations and Riboflavin-Responsive Exercise Intolerance. Available online: https://www.ncbi.nlm.nih.gov/pubmed/26933868 (accessed on 28 August 2018).
- Olsen, R.K.J.; Koňaříková, E.; Giancaspero, T.A.; Mosegaard, S.; Boczonadi, V.; Mataković, L.; Veauville-Merllié, A.; Terrile, C.; Schwarzmayr, T.; Haack, T.B.; et al. Riboflavin-Responsive and -Non-responsive mutations in FAD synthase cause multiple Acyl-CoA dehydrogenase and combined respiratory-chain deficiency. Am. J. Hum. Genet. 2016, 98, 1130–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzi, L.; Perseghin, G.; Tambussi, G.; Meneghini, E.; Scifo, P.; Pagliato, E.; Del Maschio, A.; Testolin, G.; Lazzarin, A. Intramyocellular lipid accumulation and reduced whole body lipid oxidation in HIV lipodystrophy. Am. J. Physiol. Endocrinol. MeTable 2003, 284, E274–E280. [Google Scholar] [CrossRef] [PubMed]
- Lindegaard, B.; Hvid, T.; Wolsk Mygind, H.; Mortensen, O.H.; Grøndal, T.; Abildgaard, J.; Gerstoft, J.; Pedersen, B.K.; Baranowski, M. Low expression of IL-18 and IL-18 receptor in human skeletal muscle is associated with systemic and intramuscular lipid metabolism-Role of HIV lipodystrophy. PLoS ONE 2018, 13, e0186755. [Google Scholar] [CrossRef]
- Urbano-Márquez, A.; Fernández-Solà, J. Effects of alcohol on skeletal and cardiac muscle. Muscle Nerve 2004, 30, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Preedy, V.R.; Ohlendieck, K.; Adachi, J.; Koll, M.; Sneddon, A.; Hunter, R.; Rajendram, R.; Mantle, D.; Peters, T.J. The importance of alcohol-induced muscle disease. J. Muscle Res. Cell. Motil. 2003, 24, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Sacanella, E.; Fernández-Solá, J.; Nicolás, J.M.; Rubin, E.; Urbano-Márquez, A. Natural history of alcoholic myopathy: A 5-year study. Alcohol. Clin. Exp. Res. 1998, 22, 2023–2028. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Solà, J.; Preedy, V.R.; Lang, C.H.; Gonzalez-Reimers, E.; Arno, M.; Lin, J.C.I.; Wiseman, H.; Zhou, S.; Emery, P.W.; Nakahara, T.; et al. Molecular and cellular events in alcohol-induced muscle disease. Alcohol. Clin. Exp. Res. 2007, 31, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.A.; Le, T.; Tong, M.; Silbermann, E.; Gundogan, F.; de la Monte, S.M. Impaired insulin/IGF signaling in experimental alcohol-related myopathy. Nutrients 2012, 4, 1058–1075. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, V.; Nowis, D.; Tinelli, M.; Malatesta, M.; Paoli, L.; Marini, M.; Manganotti, P.; Sadowski, R.; Wilczynski, G.M.; Meneghini, V.; et al. Bortezomib-Induced Muscle Toxicity in Multiple Myeloma. J. Neuropathol. Exp. Neurol. 2017, 76, 620–630. [Google Scholar] [CrossRef] [PubMed]

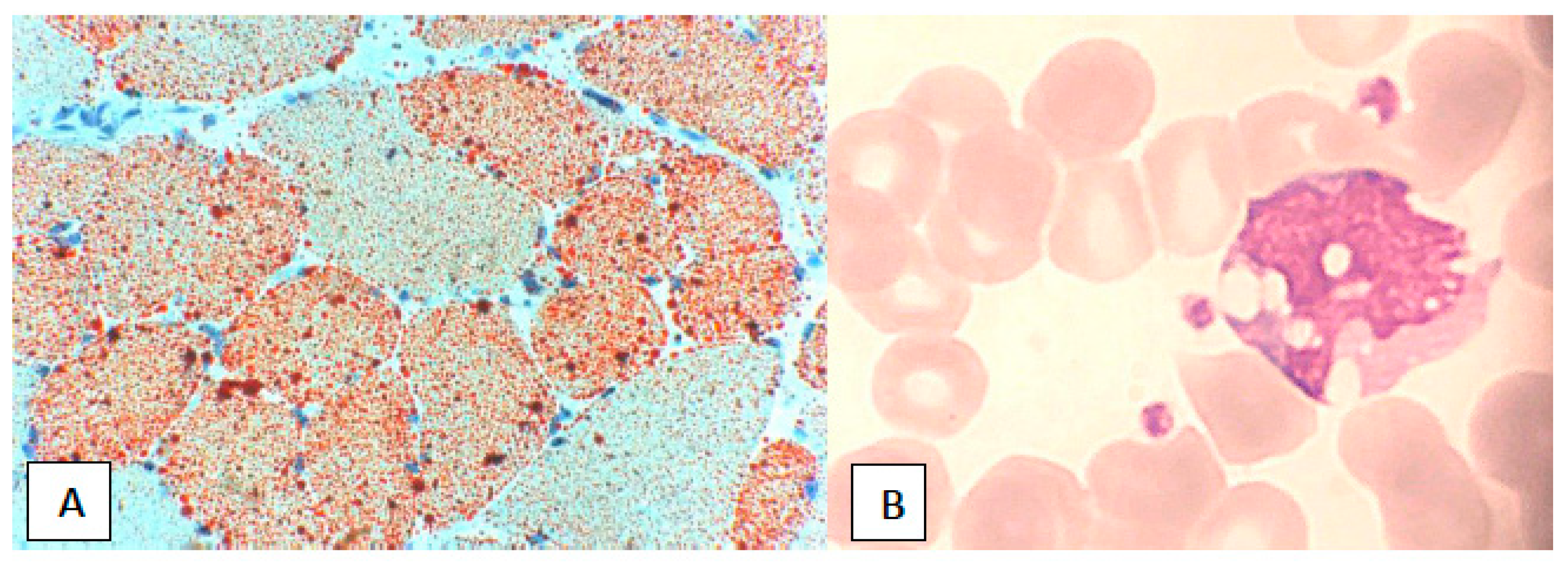

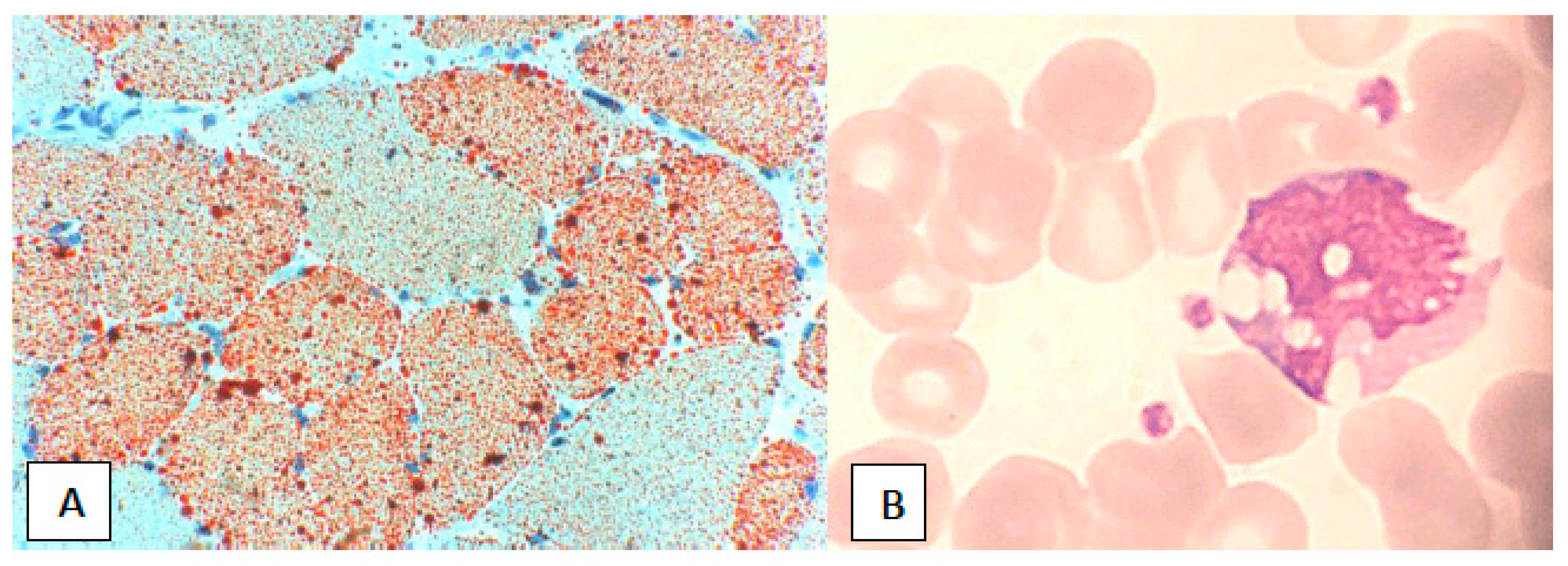
{kind=link}
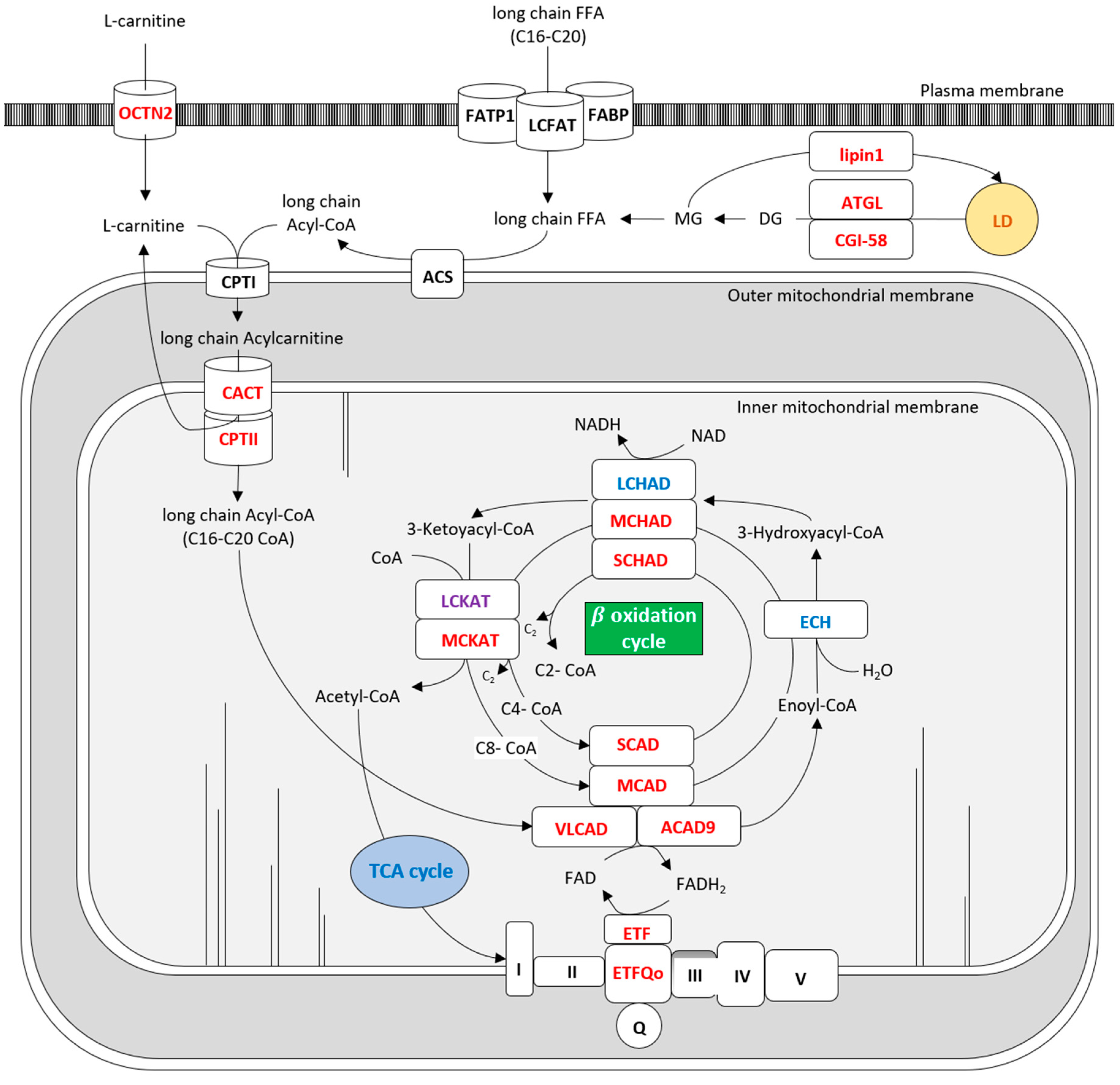
{kind=link}
| Disease Name | Gene | Protein | Predominant Symptoms | Laboratory Diagnosis | Lipid Storage | Therapy | ||
|---|---|---|---|---|---|---|---|---|
| PM | LMI | Primary Carnitine Deficiency (PCD) | SLC22A5 | OCTN2 | C, E, Hy, M, R, RL | LC, AC | +++ | Carnitine, preventing trigger factors |
| Cytoplasm | LMII | Neutral lipid storage disease type M (NLSD-M) | PNPLA2 | ATGL | C, IOL, M | JA, NC, CK | +++ | Low fat diet, MCT, exercise, fibrates, beta-adrenegic drugs |
| LMII | Neutral lipid storage disease type I (NLSD-I) | CGI58/ABHD5 | CGI58 | He, Ic, M | JA, NC | +++ | Low fat diet, MCT, carnitine, triheptanoin acid, acitrein | |
| LMIV | Phosphatidic acid phosphatase deficiency (Lipin Deficiency) | LPIN | Lipin 1 | C, He, M, R | CK, NC | +/− | Glucose and fluids infusion, monitoring of vital function | |
| LMV | Carnitine-Acylcarnitine Translocase) Deficiency (CACT) | SLC25A20 | CACT | C, E, He, Hy, M | OA, LC, AC | NR | Avoid fasting, high carbohydrate intake, MCT, polyunsaturated fatty acids, carnitine. | |
| Mitochondrion | LMVI | Carnitine palmitoyl transferase II deficiency (CPT II) | CPTII | CPTII | M, My, R, TM | AC, NC, DBS | +/− | Glucose infusion, carnitine, avoid FANS, preventing trigger factors |
| LMVII | Very-long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | ACADVL | VLCAD | C, He, R | AC | +/− | Triheptanoin acid, MCT, N-acetylcisteine, avoid fasting, | |
| LMVII | Acyl-CoA dehydrogenase 9 deficiency (ACAD9) | ACAD9 | ACAD9 | C, Ex, Hy, He, R, RL | − | Riboflavin | ||
| LMIX | Long-chain acyl-coA dehydrogenase (LCAD) | ACADL | LCAD | C, He, R | AC | +/− | Riboflavin | |
| LMX | Medium-chain acyl-CoA dehydrogenase deficiency (MCAD) | ACADM | MCAD | C, Ex, He, Hy, M, R | AC | − | Avoid fasting, glucose infusion | |
| LMXI | Short-chain acyl-CoA dehydrogenase deficiency (SCAD) | ACADS | SCAD | C, Ex, MA, M, no Hy | AC, OA | +/− | Low fat diet, carnitine, riboflavin | |
| LMXII | Mitochondrial trifunctional protein deficiency (MTP) | ADHA | ECH, LCHAD | C, E, He, M, N, NH, R, RL | AC | +/− | Decosahenoxic acid (DHA), Triheptanoin acid, PPAR | |
| ADHB | LCKAT | AC | − | |||||
| LMXIII | Short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency (SCHAD) | HADH | SCHAD | C, M, He, Hy, R | OA | +/− | No therapy reported | |
| LMXIV | Medium-chain 3-ketoacyl-CoA thiolase (MCKAT) | MCKAT | MCKAT | He, MA, R | OA, AC | NR | No therapy reported | |
| LMXV | Multiple acyl-CoA dehydrogenase deficiency (MADD) | ETFA, ETFB | ETF | C, D, Ex, He, Hy | LC, AC | +/− | Riboflavin, low fat diet, avoid fasting, CoQ | |
| ETFDH | ETFQO | C, D, Ex, He, Hy | LC, AC | +++ | Riboflavin, low fat diet, avoid fasting, CoQ |
| Genetically Determined Metabolic Errors | Acquired Medical Conditions |
|---|---|
Increased Esterification due to acyl-CoA Accumulation
| Decreased Biosynthesis
Increased Esterification and competitive inhibition of carnitine uptake by valproylcarnitine
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennisi, E.M.; Garibaldi, M.; Antonini, G. Lipid Myopathies. J. Clin. Med. 2018, 7, 472. https://doi.org/10.3390/jcm7120472
Pennisi EM, Garibaldi M, Antonini G. Lipid Myopathies. Journal of Clinical Medicine. 2018; 7(12):472. https://doi.org/10.3390/jcm7120472
Chicago/Turabian StylePennisi, Elena Maria, Matteo Garibaldi, and Giovanni Antonini. 2018. "Lipid Myopathies" Journal of Clinical Medicine 7, no. 12: 472. https://doi.org/10.3390/jcm7120472