Why, Who, When, and How? Rationale for Considering Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Why Consider Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease?
2.1. Non-Transplant Intensive Therapies in Sickle Cell Disease Patients
2.1.1. Hydroxyurea
2.1.2. Other Drugs
2.1.3. Transfusion
2.2. Stem Cell Transplantation
2.2.1. Transplantations from an HLA-Identical Sibling (Matched-Sibling Donor)
2.2.2. Unrelated Stem Cell Transplantations
2.2.3. Related Haplo-Identical Transplantations
3. Who to Consider for Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease?
3.1. Cerebral Vasculopathy
3.1.1. Physiopathology
3.1.2. Secondary Stroke Prevention
3.1.3. Primary Stroke Prevention
Cerebral Vasculopathy Detection
Primary Stroke Prevention by Chronic Transfusion
Primary Stroke Prevention by Hydroxyurea
Primary Stroke Prevention by Stem Cell Transplantation
3.1.4. Silent Cerebral Infarcts
Detection, Prevalence and Risk Factors
Prevention of Silent Cerebral Infarcts by Chronic Transfusion
Prevention of Silent Cerebral Infarcts by Hydroxyurea
Prevention of Silent Cerebral Infarcts by Stem Cell Transplantation
3.1.5. Cognitive deficiency
3.2. Prevention of Vaso-Occlusive Crisis and Acute Chest Syndrome
3.3. Spleen Function
3.4. Kidney Function
3.5. Pulmonary Function
3.6. Gonadal Function
3.7. Growth
3.8. Osteonecrosis
3.9. Health-Related Quality of Life
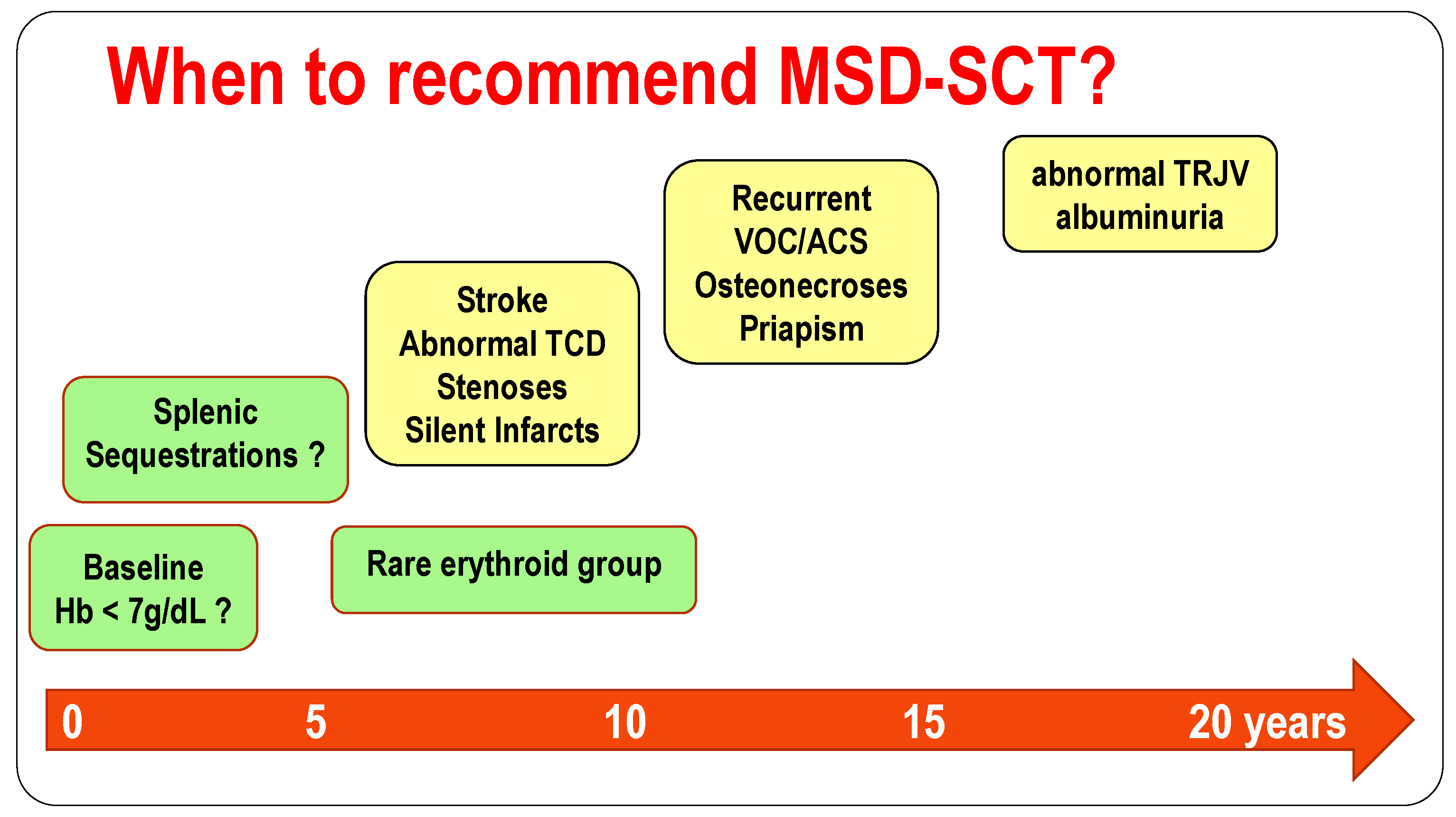
4. When to Consider Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease?
5. How? Choice of Conditioning Regimen and Transplantation Types as a Function of Donor Availability and Disease State
5.1. Cerebral Vasculopathy
5.2. Crises Vaso-Occlusives (CVO) and/or Acute Chest Syndromes (ACS)
5.3. Children with MSD: Choice between Myeloablative or Non-Myeloablative Conditioning Regimen?
5.4. Children without MSD: Choice between Unrelated Transplantation or Related-Haplo-Identical?
5.5. Children without MSD: is Gene Therapy in the Near Future?
6. Summary
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global burden of sickle cell anaemia in children under five, 2010–2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Gaston, M.H.; Verter, J.I.; Woods, G.; Pegelow, C.; Kelleher, J.; Presbury, G.; Zarkowsky, H.; Vichinsky, E.; Iyer, R.; Lobel, J.S.; et al. Prophylaxis with oral penicillin in children with sickle cell anemia. N. Engl. J. Med. 1986, 314, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Adamkiewicz, T.V.; Silk, B.J.; Howgate J Baughman, W.; Strayhorn, G.; Sullivan, K.; Farley, M.M. Effectiveness of the 7-valent pneumococcal conjugate vaccine in children with sickle cell disease in the first decade of life. Pediatrics 2008, 121, 562–569. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.V.; McMahon, R.P.; Bonds, D.R. Effect of hydroxyurea on the frequency of painful crisis in sickle cell anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Robinson, S.; Howard, J. How I manage red cell transfusions in patients with sickle cell disease. Br. J. Haematol. 2018, 180, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Vichinsky, E.; Bernaudin, F.; Forni, G.L.; Gardner, R.; Hassell, K.; Heeney, M.M.; Inusa, B.; Kutlar, A.; Lane, P.; Mathias, L.; et al. Long-term safety and efficacy of deferasirox (Exjade) for up to 5 years in transfusional iron-overloaded patients with sickle cell disease. Br. J. Haematol. 2011, 154, 387–397. [Google Scholar] [CrossRef]
- Adams, R.J.; McKie, V.; Nichols, F.; Carl, E.; Zhang, D.L.; McKie, K.; Figueroa, R.; Litaker, M.; Thompson, W.; Hess, D. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N. Engl. J. Med. 1992, 326, 605–610. [Google Scholar] [CrossRef]
- Verlhac, S.; Bernaudin, F.; Tortrat D Brugieres, P.; Mage, K.; Gaston, A.; Reinert, P. Detection of cerebrovascular disease in sickle cell disease children by transcranial Doppler sonography. Correlation with MRI and MRA and conventional angiography. Pediatric Radiol. 1995, 25, S14–S19. [Google Scholar]
- Vermylen, C.; Fernandez Robles, E.; Ninane, J.; Cornu, G. Bone marrow transplantation in five children with sickle cell anaemia. Lancet 1988, 1, 1427–1428. [Google Scholar] [CrossRef]
- Bernaudin, F.; Souillet, G.; Vannier, J.P.; Plouvier, E.; Lemerle, S.; Michel, G.; Bordigoni, P.; Lutz, P.; Kuentz, M. Bone marrow transplantation (BMT) in 14 children with severe sickle cell disease (SCD): The French experience. GEGMO. Bone Marrow Transplant. 1993, 12 (Suppl. S1), 118–121. [Google Scholar] [PubMed]
- Walters, M.C.; Patience, M.; Leisenring, W.; Eckman, J.R.; Scott, J.P.; Mentzer, W.C.; Davies, S.C.; Ohene-Frempong, K.; Bernaudin, F.; Matthews, D.C.; et al. Bone marrow transplantation for sickle cell disease. N. Engl. J. Med. 1996, 335, 369–376. [Google Scholar] [CrossRef]
- Walters, M.C.; Storb, R.; Patience, M.; Leisenring, W.; Taylor, T.; Sanders, J.E.; Buchanan, G.E.; Rogers, Z.R.; Dinndorf, P.; Davies, S.C.; et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: An interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood 2000, 95, 1918–1924. [Google Scholar]
- Bernaudin, F.; Socie, G.; Kuentz, M.; Chevret, S.; Duval, M.; Bertrand, Y.; Vannier, J.P.; Yakouben, K.; Thuret, I.; Bordigoni, P.; et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007, 110, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Pinto Simões, B.; Ferster, A.; Dupont, S.; de la Fuente, J.; et al. Eurocord, the Pediatric Working Party of the European Society for Blood and Marrow Transplantation, and the Center for International Blood andMarrow Transplant Research. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar]
- Bernaudin, F.; Dalle, J.H.; Bories, D.; Peffault de Latour, R.; Robin, M.; Bertrand, Y.; Pondarre, C.; Vannier, J.P.; Neven, B.; Kuentz, M.; et al. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica 2018, pii: Haematol.2018.213207. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Kang, E.M.; Fitzhugh, C.D.; Link, M.B.; Bolan, C.D.; Kurlander, R.; Childs, R.W.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N. Engl. J. Med. 2009, 361, 2309–2317. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Fitzhugh, C.D.; Weitzel, R.P.; Link, M.E.; Coles, W.A.; Zhao, X.; Rodgers, G.P.; Powell, J.D.; Tisdale, J.F. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014, 312, 48–56. [Google Scholar] [CrossRef]
- Saraf, S.L.; Oh, A.L.; Patel, P.R.; Jalundhwala, Y.; Sweiss, K.; Koshy, M.; Campbell-Lee, S.; Gowhari, M.; Hassan, J.; Peace, D.; et al. Nonmyeloablative Stem Cell Transplantation with Alemtuzumab/Low-Dose Irradiation to Cure and Improve the Quality of Life of Adults with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2016, 22, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Bolaños-Meade, J.; Fuchs, E.J.; Luznik, L.; Lanzkron, S.M.; Gamper, C.J.; Jones, R.J.; Brodsky, R.A. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 2012, 120, 4285–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamani, N.R.; Walters, M.C.; Carter, S.; Aquino, V.; Brochstein, J.A.; Chaudhury, S.; Eapen, M.; Freed, B.M.; Grimley, M.; Levine, J.E.; et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: Results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol. Blood Marrow Transplant. 2012, 18, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.; Eapen, M.; Panepinto, J.A.; Logan, B.R.; Wu, J.; Abraham, A.; Brochstein, J.; Chaudhury, S.; Godder, K.; Haight, A.E.; et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood 2016, 128, 2561–2567. [Google Scholar] [CrossRef]
- Poirot, C.; Brugieres, L.; Yakouben, K.; Prades-Borio, M.; Marzouk, F.; de Lambert, G.; Pacquement, H.; Bernaudin, F.; Neven, B.; Paye-Jaouen, A.; et al. Ovarian tissue cryopreservation for fertility preservation in 418 girls and adolescents up to 15 years of age facing highly gonadotoxic treatment. Twenty years of experience at a single center. Acta Obstet. Gynecol. Scand. 2019, 98, 630–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stukenborg, J.B.; Alves-Lopes, J.P.; Kurek, M.; Albalushi, H.; Reda, A.; Keros, V.; Töhönen, V.; Bjarnason, R.; Romerius, P.; Sundin, M.; et al. Spermatogonial quantity in human prepubertal testicular tissue collected for fertility preservation prior to potentially sterilizing therapy. Hum. Reprod. 2018, 33, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.T.; Rogers, Z.R.; Buchanan, G.R. Survival of children with sickle cell disease. Blood 2004, 103, 4023–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telfer, P.; Coen, P.; Chakravorty, S.; Wilkey, O.; Evans, J.; Newell, H.; Smalling, B.; Amos, R.; Stephens, A.; Rogers, D.; et al. Clinical outcomes in children with sickle cell disease living in England: A neonatal cohort in East London. Haematologica 2007, 92, 905–912. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Arnaud, C.; Kamdem, A.; Chevret, S.; Hau, I.; Coïc, L.; Leveillé, E.; Lemarchand, E.; Lesprit, E.; et al. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 2011, 117, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Quinn, C.T.; Rogers, Z.R.; McCavit, T.L.; Buchanan, G.R. Improved survival of children and adolescents with sickle cell disease. Blood 2010, 115, 3447–3452. [Google Scholar] [CrossRef]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Lanzkron, S.; Carroll, C.P.; Haywood, C., Jr. Mortality rates and age at death from sickle cell disease: U.S.; 1979–2005. Public Health Rep. 2013, 128, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Grosse, S.D.; Odame, I.; Atrash, H.K.; Amendah, D.D.; Piel, F.B.; Williams, T.N. Sickle cell disease in Africa: A neglected cause of early childhood mortality. Am. J. Prev. Med. 2011, 41 (Suppl. S4), 398–405. [Google Scholar] [CrossRef] [PubMed]
- Charache, S.; Dover, G.J.; Moore, R.D.; Eckert, S.; Ballas, S.K.; Koshy, M.; Milner, P.F.; Orringer, E.P.; Phillips, G., Jr.; Platt, O.S.; et al. Hydroxyurea: Effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992, 79, 2555–2565. [Google Scholar] [PubMed]
- Ferster, A.; Vermylen, C.; Cornu, G.; Buyse, M.; Corazza, F.; Devalck, C.; Fondu, P.; Toppet, M.; Sariban, E. Hydroxyurea for treatment of severe sickle cell anemia: A pediatric clinical trial. Blood 1996, 88, 1960–1964. [Google Scholar] [PubMed]
- De Montalembert, M.; Belloy, M.; Bernaudin, F.; Gouraud, F.; Capdeville, R.; Mardini, R.; Philippe, N.; Jais, J.P.; Bardakdjian, J.; Ducrocq, R.; et al. Three-year follow-up of hydroxyurea treatment in severely ill children with sickle cell disease. The French Study Group on Sickle Cell Disease. J. Pediatr Hematol./Oncol. 1997, 19, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Ware, R.E.; Miller, S.T.; Iyer, R.V.; Casella, J.F.; Minniti, C.P.; Rana, S.; Thornburg, C.D.; Rogers, Z.R.; Kalpatthi, R.V.; et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (BABY HUG). Lancet 2011, 377, 1663–1672. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Barton, F.; Castro, O.; Pegelow, C.H.; Ballas, S.K.; Kutlar, A.; Orringer, E.; Bellevue, R.; Olivieri, N.; Eckman, J.; et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: Risks and benefits up to 9 years of treatment. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef]
- Steinberg, M.H.; McCarthy, W.F.; Castro, O.; Ballas, S.K.; Armstrong, F.D.; Smith, W.; Ataga, K.; Swerdlow, P.; Kutlar, A.; DeCastro, L.; et al. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia and MSH Patients’ Follow-Up. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am. J. Hematol. 2010, 85, 403–408. [Google Scholar]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: Results of a 17-year, single-center trial (LaSHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Hsieh, M.M.; Allen, D.; Coles, W.A.; Seamon, C.; Ring, M.; Zhao, X.; Minniti, C.P.; Rodgers, G.P.; Schechter, A.N.; et al. Hydroxyurea-Increased Fetal Hemoglobin Is Associated with Less Organ Damage and Longer Survival in Adults with Sickle Cell Anemia. PLoS ONE 2015, 10, e0141706. [Google Scholar] [CrossRef]
- Bernaudin, F.; Arnaud, C.; Kamdem, A.; Hau, I.; Lelong, F.; Epaud, R.; Pondarré, C.; Pissard, S. Biological impact of α genes, β haplotypes, and G6PD activity in sickle cell anemia at baseline and with hydroxyurea. Blood Adv. 2018, 2, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Campbell, A.; Rana, S.; Nouraie, M.; Niu, X.; Minniti, C.P.; Sable, C.; Darbari, D.; Dham, N.; Onyekwere, O.; et al. Relationship of erythropoietin, fetal hemoglobin, and hydroxyurea treatment to tricuspid regurgitation velocity in children with sickle cell disease. Blood 2009, 114, 4639–4644. [Google Scholar] [CrossRef] [PubMed]
- Rigano, P.; Pecoraro, A.; Calvaruso, G.; Steinberg, M.H.; Iannello, S.; Maggio, A. Cerebrovascular events in sickle cell-beta thalassemia treated with hydroxyurea: A single center prospective survey in adult Italians. Am. J. Hematol. 2013, 88, E261–E264. [Google Scholar] [CrossRef]
- DeBaun, M.R. Hydroxyurea therapy contributes to infertility in adult men with sickle cell disease: A review. Expert Rev. Hematol. 2014, 7, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Yawn, B.P.; Buchanan, G.R.; Afenyi-Annan, A.N.; Ware, R.E.; Murad, H.; Ortiz, E.; Fulwood, R.; Horton, A.; John-Sowah, J.; Savage, W.J.; et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA 2014, 312, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Niihara, Y.; Smith, W.R.; Stark, C.W. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 1880. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Kutlar, A.; Kanter, J. Crizanlizumab in Sickle Cell Disease. N. Engl. J. Med. 2017, 4, 1796. [Google Scholar]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Yazdanbakhsh, K.; Ware, R.E.; Noizat-Pirenne, F. Red blood cell alloimmunization in sickle cell disease: Pathophysiology, risk factors, and transfusion management. Blood 2012, 120, 528–537. [Google Scholar] [CrossRef]
- Wayne, A.S.; Schoenike, S.E.; Pegelow, C.H. Financial analysis of chronic transfusion for stroke prevention in sickle cell disease. Blood 2000, 96, 2369–2372. [Google Scholar]
- Lo, W.; Zamel, K.; Ponnappa, K.; Allen, A.; Chisolm, D.; Tang, M.; Kerlin, B.; Yeates, K.O. The Cost of Pediatric Stroke Care and Rehabilitation. Stroke 2008, 39, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Guilcher, G.M.T.; Truong, T.H.; Saraf, S.L.; Joseph, J.J.; Rondelli, D.; Hsieh, M.M. Curative therapies: Allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and nonmyeloablative conditioning in sickle cell disease. Semin Hematol. 2018, 55, 87–93. [Google Scholar] [CrossRef]
- Bernaudin, F.; Kuentz, M. Haplo-BMT: Cure or back to sickle cell? Blood 2012, 120, 4276–4277. [Google Scholar] [CrossRef]
- De la Fuente, J.; Dhedin, N.; Koyama, T.; Bernaudin, F.; Kuentz, M.; Karnik, L.; Socié, G.; Culos, K.A.; Brodsky, R.A.; DeBaun, M.R.; et al. Haploidentical Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide Plus Thiotepa Improves Donor Engraftment in Patients with Sickle Cell Anemia: Results of an International Learning Collaborative. Biol. Blood Marrow Transplant. 2019, 25, 1197–1209. [Google Scholar] [CrossRef]
- Foell, J.; Pfirstinger, B.; Rehe, K.; Wolff, D.; Holler, E.; Corbacioglu, S. Haploidentical stem cell transplantation with CD3(+)-/CD19(+)- depleted peripheral stem cells for patients with advanced stage sickle cell disease and noalternative donor: Results of a pilot study. Bone Marrow Transplant. 2017, 52, 938–940. [Google Scholar] [CrossRef]
- Connes, P.; Verlhac, S.; Bernaudin, F. Advances in understanding the pathogenesis of cerebrovascular vasculopathy in sickle cell anaemia. Br. J. Haematol. 2013, 161, 484–498. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Chevret, S.; Torres, M.; Coic, L.; Arnaud, C.; Kamdem, A.; Hau, I.; Neonato, M.G.; Delacourt, C. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood 2008, 112, 4314–4317. [Google Scholar] [CrossRef]
- Ohene-Frempong, K.; Weiner, S.J.; Sleeper, L.A.; Miller, S.T.; Embury, S.; Moohr, J.W.; Wethers, D.L.; Pegelow, C.H.; Gill, F.M. Cerebrovascular accidents in sickle cell disease: Rates and risk factors. Blood 1998, 91, 288–294. [Google Scholar]
- Russell, M.O.; Goldberg, H.I.; Hodson, A.; Kim, H.C.; Halus, J.; Reivich, M.; Schwartz, E. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood 1984, 63, 162–169. [Google Scholar] [Green Version]
- Wang, W.C.; Kovnar, E.H.; Tonkin, I.L.; Mulhern, R.K.; Langston, J.W.; Day, S.W.; Schell, M.J.; Wilimas, J.A. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. J. Pediatrics 1991, 118, 377–382. [Google Scholar] [CrossRef]
- Hulbert, M.L.; McKinstry, R.C.; Lacey, J.L.; Moran, C.J.; Panepinto, J.A.; Thompson, A.A.; Sarnaik, S.A.; Woods, G.M.; Casella, J.F.; Inusa, B.; et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood 2011, 117, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Brousse, V.; Hertz-Pannier, L.; Consigny, Y. Does regular blood transfusion prevent progression of cerebrovascular lesions in children with sickle cell disease? Ann. Hematol. 2009, 88, 785–788. [Google Scholar] [CrossRef]
- Bader-Meunier, B.; Verlhac, S.; Elmaleh-Bergès, M.; Ithier, G.; Sellami, F.; Faid, S.; Missud, F.; Ducrocq, R.; Alberti, C.; Zaccaria, I.; et al. Effect of transfusion therapy on cerebral vasculopathy in children with sickle-cell anemia. Haematologica 2009, 94, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Ware, R.E.; Zimmerman, S.A.; Schultz, W.H. Hydroxyurea as an alternative to blood transfusions for the prevention of recurrent stroke in children with sickle cell disease. Blood 1999, 94, 3022–3026. [Google Scholar]
- Ware, R.E.; Helms, R.W. SWiTCH Investigators. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH). Blood 2012, 119, 3925–3932. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Arnaud, C.; Kamdem, A.; Hau, I.; Leveillé, E.; Vasile, M.; Kasbi, F.; Madhi, F.; Fourmaux, C.; et al. Long-term treatment follow-up of children with sickle cell disease monitored with abnormal transcranial Doppler velocities. Blood 2016, 127, 1814–1822. [Google Scholar] [CrossRef]
- Vermylen, C.; Cornu, G.; Ferster, A.; Brichard, B.; Ninane, J.; Ferrant, A.; Zenebergh, A.; Maes, P.; Dhooge, C.; Benoit, Y.; et al. Haematopoietic stem cell transplantation for sickle cell anaemia: The first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998, 22, 1–6. [Google Scholar] [CrossRef]
- Dobson, S.R.; Holden, K.R.; Nietert, P.J.; Cure, J.K.; Laver, J.H.; Disco, D.; Abboud, M.R. Moyamoya syndrome in childhood sickle cell disease: A predictive factor for recurrent cerebrovascular events. Blood 2002, 99, 3144–3150. [Google Scholar] [CrossRef]
- Calviere, L.; Viguier, A.; Guidolin, B.; Tall, P.; Larrue, V. Cervical artery stenoses in sickle cell disease. Eur. Neurol. 2007, 58, 120–121. [Google Scholar] [CrossRef]
- Gorman, M.J.; Nystrom, K.; Carbonella, J.; Pearson, H. Submandibular TCD approach detects post-bulb ICA stenosis in children with sickle cell anemia. Neurology 2009, 73, 362–365. [Google Scholar] [CrossRef]
- Deane, C.R.; Goss, D.; Bartram, J.; Pohl, K.R.; Height, S.E.; Sibtain, N.; Jarosz, J.; Thein, S.L.; Rees, D.C. Extracranial internal carotid arterial disease in children with sickle cell anemia. Haematologica 2010, 95, 1287–1292. [Google Scholar] [CrossRef]
- Telfer, P.T.; Evanson, J.; Butler, P.; Hemmaway, C.; Abdulla, C.; Gadong, N.; Whitmarsh, S.; Kaya, B.; Kirkham, F.J. Cervical carotid artery disease in sickle cell disease: Clinical and radiological features. Blood 2011, 118, 6192–6199. [Google Scholar] [CrossRef]
- Verlhac, S.; Balandra, S.; Cussenot, I.; Kasbi, F.; Vasile, M.; Kheniche, A.; Elmaleh-Bergès, M.; Ithier, G.; Benkerrou, M.; Bernaudin, F.; et al. Extracranial carotid arteriopathy in stroke-free children with sickle cell anemia: Detection by submandibular Doppler sonography. Pediatric Radiol. 2014, 44, 587–596. [Google Scholar] [CrossRef]
- Adams, R.J.; McKie, V.C.; Carl, E.M.; Nichols, F.T.; Perry, R.; Brock, K.; McKie, K.; Figueroa, R.; Litaker, M.; Weiner, S.; et al. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann. Neurol. 1997, 42, 699–704. [Google Scholar] [CrossRef]
- Adams, R.J.; McKie, V.C.; Hsu, L.; Files, B.; Vichinsky, E.; Pegelow, C.; Abboud, M.; Gallagher, D.; Kutlar, A.; Nichols, F.T.; et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med. 1998, 339, 5–11. [Google Scholar] [CrossRef]
- Adams, R.J. Brambilla D of the STOP 2 investigative team. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N. Engl. J. Med. 2005, 353, 2769–2778. [Google Scholar]
- Bernaudin, F.; Verlhac, S.; Coïc, L.; Lesprit, E.; Brugières, P.; Reinert, P. Long term follow-up of pediatric sickle cell disease patients with abnormal high velocities on transcranial Doppler. Pediatric Radiol. 2005, 35, 242–248. [Google Scholar] [CrossRef]
- Ware, R.E.; Davis, B.R.; Schultz WH Brown, R.C.; Aygun, B.; Sarnaik, S.; Odame, I.; Fuh, B.; George, A.; Owen, W.; Luchtman-Jones, L.; et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD with Transfusions Changing to Hydroxyurea (TWiTCH): A multicentre, open-label, phase 3, non-inferiority trial. Lancet 2016, 387, 661–670. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Chevret, S. Treating sickle cell anaemia: The TWiTCH trial. Lancet 2016, 388, 960. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Peffault de Latour, R.; Dalle, J.H.; Brousse, V.; Petras, E.; Thuret, I.; Paillard, C.; Neven, B.; Galambrun, C.; et al. Association of Matched Sibling Donor Hematopoietic Stem Cell Transplantation With Transcranial Doppler Velocities in Children With Sickle Cell Anemia. JAMA 2019, 321, 266–276. [Google Scholar] [CrossRef]
- Armstrong, F.D.; Thompson, R.J., Jr.; Wang, W.; Zimmerman, R.; Pegelow, C.H.; Miller, S.; Moser, F.; Bello, J.; Hurtig, A.; Vass, K. Cognitive functioning and brain magnetic resonance imaging in children with sickle Cell disease. Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Pediatrics 1996, 97, 864–870. [Google Scholar]
- Bernaudin, F.; Verlhac, S.; Fréard, F.; Roudot-Thoraval, F.; Benkerrou, M.; Thuret, I.; Mardini, R.; Vannier, J.P.; Ploix, E.; Romero, M.; et al. Multicenter prospective study of children with sickle cell disease: Radiographic and psychometric correlation. J. Child. Neurol. 2000, 15, 333–343. [Google Scholar] [CrossRef]
- Schatz, J.; Brown, R.T.; Pascual, J.M.; Hsu, L.; DeBaun, M.R. Poor school and cognitive functioning with silent cerebral infarcts and sickle cell disease. Neurology 2001, 56, 1109–1111. [Google Scholar] [CrossRef] [Green Version]
- Pegelow, C.H.; Macklin, E.A.; Moser, F.G.; Wang, W.C.; Bello, J.A.; Miller, S.T.; Vichinsky, E.P.; DeBaun, M.R.; Guarini, L.; Zimmerman, R.A.; et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood 2002, 99, 3014–3018. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.T.; Macklin, E.A.; Pegelow, C.H.; Kinney, T.R.; Sleeper, L.A.; Bello, J.A.; DeWitt, L.D.; Gallagher, D.M.; Guarini, L.; Moser, F.G.; et al. Silent infarction as a risk factor for overt stroke inchildren with sickle cell anemia: A report from the Cooperative Study of Sickle Cell Disease. J. Pediatrcs 2001, 139, 385–390. [Google Scholar] [CrossRef]
- Kassim, A.A.; Pruthi, S.; Day, M.; Rodeghier, M.; Gindville, M.C.; Brodsky, M.A.; DeBaun, M.R.; Jordan, L.C. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood 2016, 127, 2038–2040. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Sarnaik, S.A.; Rodeghier, M.J.; Minniti, C.P.; Howard, T.H.; Iyer, R.V.; Inusa, B.; Telfer, P.T.; Kirby-Allen, M.; Quinn, C.T.; et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: Low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 2012, 119, 3684–3690. [Google Scholar] [CrossRef]
- Kwiatkowski, J.L.; Zimmerman, R.A.; Pollock, A.N.; Seto, W.; Smith-Whitley, K.; Shults, J.; Blackwood-Chirchir, A.; Ohene-Frempong, K. Silent infarcts in young children with sickle cell disease. Br. J. Haematol. 2009, 146, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Bernaudin, F.; Verlhac, S.; Arnaud, C.; Kamdem, A.; Vasile, M.; Kasbi, F.; Hau, I.; Madhi, F.; Fourmaux, C.; Biscardi, S.; et al. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood 2015, 125, 1653–1661. [Google Scholar] [CrossRef] [Green Version]
- DeBaun, M.R.; Gordon, M.; McKinstry, R.C.; Noetzel, M.J.; White, D.A.; Sarnaik, S.A.; Meier, E.R.; Howard, T.H.; Majumdar, S.; Inusa, B.P.; et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N. Engl. J. Med. 2014, 371, 699–710. [Google Scholar] [CrossRef]
- Nottage, K.A.; Ware, R.E.; Aygun, B.; Smeltzer, M.; Kang, G.; Moen, J.; Wang, W.C.; Hankins, J.S.; Helton, K.J. Hydroxycarbamide treatment and brain MRI/MRA findings in children with sickle cell anaemia. Br. J. Haematol. 2016, 175, 331–338. [Google Scholar] [CrossRef]
- Rushton, T.; Aban, I.; Young, D.; Howard, T.; Hilliard, L.; Lebensburger, J. Hydroxycarbamide for patients with silent cerebral infarcts: Outcomes and patient preference. Br. J. Haematol. 2018, 181, 145–148. [Google Scholar] [CrossRef]
- Amstrong, F.D.; Thompson, R.J.; Wang, W.; Zimmerman, R.; Pegelow, C.H.; Miller, S.; Moser, F.; Bello, J.; Hurtig, A. Kerstin Vass for the Neuropsychology Committee of the Cooperative Study of Sickle Cell Diseas. Cognitive functioning and brain magnetic resonance imaging in children with sickle cell disease. Pediatrics 1996, 97, 864–870. [Google Scholar]
- DeBaun, M.R.; Schatz, J.; Siegel, M.J.; Koby, M.; Craft, S.; Resar, L.; Chu, J.-Y.; Launius, G.; Dadash-Zadeh, M.; Lee, R.B.; et al. Cognitive screening exam-inations for silent cerebral infarcts in sickle cell disease. Neurology 1998, 50, 1678–1682. [Google Scholar] [CrossRef]
- Colombatti, R.; Lucchetta, M.; Montanaro, M.; Rampazzo, P.; Ermani, M.; Talenti, G.; Baracchini, C.; Favero, A.; Basso, G.; Manara, R.; et al. Cognition and the Default Mode Network in Children with Sickle Cell Disease: A Resting State Functional MRI Study. PLoS ONE 2016, 11, e0157090. [Google Scholar] [CrossRef]
- Kawadler, J.M.; Clayden, J.D.; Clark, C.A.; Kirkham, F.J. Intelligence quotient in paediatric sickle cell disease: A systematic review and meta-analysis. Dev. Med. Child. Neurol. 2016, 58, 672–679. [Google Scholar] [CrossRef]
- Vichinsky, E.P.; Neumayr, L.D.; Gold, J.I.; Weiner, M.W.; Rule, R.R.; Truran, D.; Kasten, J.; Eggleston, B.; Kesler, K.; McMahon, L.; et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA 2010, 303, 1823–1831. [Google Scholar] [CrossRef]
- Wang, W.; Enos, L.; Gallagher, D.; Thompson, R.; Guarini, L.; Vichinsky, E.; Wright, E.; Zimmerman, R.; Armstrong, F.D. Cooperative Study of Sickle Cell Disease. Neuropsychologic performance in school-aged children with sickle cell disease: A report from the Cooperative Study of Sickle Cell Disease. J. Pediatrics 2001, 139, 391–397. [Google Scholar] [CrossRef]
- Lebensburger, J.D.; Miller, S.T.; Howard, T.H.; Casella, J.F.; Brown, R.C.; Lu, M.; Iyer, R.V.; Sarnaik, S.; Rogers, Z.R.; Wang, W.C. BABY HUG Investigators. Influence of severity of anemia on clinical findings in infants with sickle cell anemia: Analyses from the BABY HUG study. Pediatr. Blood Cancer. 2012, 59, 675–678. [Google Scholar] [CrossRef]
- King, A.A.; McKinstry, R.C.; Wu, J.; Eapen, M.; Abel, R.; Varughese, T.; Kamani, N.; Shenoy, S. Functional and Radiologic Assessment of the Brain after Reduced-Intensity Unrelated Donor Transplantation for Severe Sickle Cell Disease: Blood and Marrow Transplant Clinical Trials Network Study 0601. Biol. Blood Marrow Transplant. 2019, 25, e174–e178. [Google Scholar] [CrossRef]
- Thornburg, C.D.; Files, B.A.; Luo, Z.; Miller, S.T.; Kalpatthi, R.; Iyer, R.; Seaman, P.; Lebensburger, J.; Alvarez, O.; Thompson, B.; et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012, 120, 4304–4310. [Google Scholar] [CrossRef] [Green Version]
- Pearson, H.A.; Cornelius, E.A.; Schwartz, A.D.; Zelson, J.H.; Wolfson, S.L.; Spencer, R.P. Transfusion-reversible functional asplenia in young children with sickle-cell anemia. N. Engl. J. Med. 1970, 283, 334. [Google Scholar] [CrossRef]
- Nottage, K.A.; Ware, R.E.; Winter, B.; Smeltzer, M.; Wang, W.C.; Hankins, J.S.; Dertinger, S.D.; Shulkin, B.; Aygun, B. Predictors of splenic function preservation in children with sickle cell anemia treated with hydroxyurea. Eur. J. Haematol. 2014, 93, 377–383. [Google Scholar] [CrossRef]
- Ferster, A.; Bujan, W.; Corazza, F.; Devalck, C.; Fondu, P.; Toppet, M.; Sariban, E.; Verhas, M. Bone marrow transplantation corrects the splenic reticuloendothelial dysfunction in sickle cell anemia. Blood 1993, 81, 1102–1105. [Google Scholar] [Green Version]
- Nickel, R.S.; Seashore, E.; Lane, P.A.; Alazraki, A.L.; Horan, J.T.; Bhatia, M.; Haight, A.E. Improved Splenic Function After Hematopoietic Stem Cell Transplant for Sickle Cell Disease. Pediatric Blood Cancer 2016, 63, 908–913. [Google Scholar] [CrossRef]
- Nath, K.A.; Hebbel, R.P. Sickle cell disease: Renal manifestations and mechanisms. Nat. Rev. Nephrol. 2015, 11, 161–171. [Google Scholar] [CrossRef]
- Sharpe, C.C.; Thein, S.L. Sickle cell nephropathy—A practical ap- proach. Br. J. Haematol. 2011, 155, 287–297. [Google Scholar] [CrossRef]
- Wigfall, D.R.; Ware, R.E.; Burchinal, M.R.; Kinney, T.R.; Foreman, J.W. Prevalence and clinical correlates of glomerulopathy in children with sickle cell disease. J. Pediatr. 2000, 136, 749–753. [Google Scholar] [CrossRef]
- Ataga, K.I.; Derebail, V.K.; Archer, D.R. The glomerulopathy of sickle cell disease. Am. J. Hematol. 2014, 89, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Bartolucci, P.; Habibi, A.; Stehle, T.; Grimbert, P.; Rakotoson, M.G.; Gellen-Dautremer, J.; Loric, S.; Moutereau, S.; Sahali, D.; Wagner-Ballon, O.; et al. Six months of hydroxyurea reduces albuminuria in patients with sickle cell disease. J. Am. Soc. Nephrol. 2016, 27, 1847–1853. [Google Scholar] [CrossRef]
- Klings, E.S.; Wyszynski, D.F.; Nolan, V.G.; Steinberg, M.H. Abnormal pulmonary function in adults with sickle cell anemia. Am. J. Respir. Crit. Care Med. 2006, 173, 1264–1269. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Perl, S.; Hsieh, M.M. Late effects of myeloablative bone marrow transplantation (BMT) in sickle cell disease (SCD). Blood 2008, 111, 1742–1743. [Google Scholar] [CrossRef] [Green Version]
- Bernaudin, F.; Kuentz, M.; Socié, G. Response: Late effects of myeloablative stem cell transplantation or late effects of sickle cell disease itself? Blood 2008, 111, 1744. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med. 2004, 350, 886–895. [Google Scholar] [CrossRef]
- Osegbe, D.N.; Akinyanju, O.; Amaku, E.O. Fertility in males with sickle cell disease. Lancet 1981, 2, 275–276. [Google Scholar] [CrossRef]
- Agbaraji, V.O.; Scott, R.B.; Leto, S.; Kingslow, L.W. Fertility studies in sickle cell disease: Semen analysis in adult male patients. Int. J. Fertil. 1988, 33, 347–352. [Google Scholar]
- Hernigou, P.; Daltro, G.; Filippini, P.; Mukasa, M.M.; Manicom, O. Percutaneous implantation of autologous bone marrow osteoprogenitor cells as treatment of bone avascular necrosis related to sickle cell disease. Open Orthop. J. 2008, 2, 62–65. [Google Scholar] [CrossRef]
- Novais, E.N.; Sankar, W.N.; Wells, L.; Carry, P.M.; Kim, Y.J. Preliminary Results of Multiple Epiphyseal Drilling and Autologous Bone Marrow Implantation for Osteonecrosis of the Femoral Head Secondary to Sickle Cell Disease in Children. J. Pediatric Orthop. 2015, 35, 810–815. [Google Scholar] [CrossRef]
- Hernigou, P.; Bernaudin, F.; Reinert, P.; Kuentz, M.; Vernant, J.P. Favorable evolution of Sickle Cell Disease (SCD) related osteonecrosis after bone marrow transplantation. J. Bone Jt. Surg. 1997, 79, 1726–1730. [Google Scholar] [CrossRef]
- Panepinto, J.A. Health-related quality of life in patients with hemoglobinopathies. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 284–289. [Google Scholar]
- Ballas, S.K.; Barton, F.B.; Waclawiw, M.A.; Swerdlow, P.; Eckman, J.R.; Pegelow, C.H.; Koshy, M.; Barton, B.A.; Bonds, D.R. Hydroxyurea and sickle cell anemia: Effect on quality of life. Health Qual. Life Outcomes 2006, 4, 59. [Google Scholar] [CrossRef]
- Thornburg, C.D.; Calatroni, A.; Panepinto, J.A. Differences in health-related quality of life in children with sickle cell disease receiving hydroxyurea. J. Pediatric Hematol. Oncol. 2011, 33, 251–254. [Google Scholar] [CrossRef]
- Badawy, S.M.; Thompson, A.A.; Lai, J.S.; Penedo, F.J.; Rychlik, K.; Liem, R.I. Health-related quality of life and adherence to hydroxyurea in adolescents and young adults with sickle cell disease. Pediatric Blood Cancer 2017, 64, e26369. [Google Scholar] [CrossRef]
- Beverung, L.M.; Strouse, J.J.; Hulbert, M.L.; Neville, K.; Liem, R.I.; Inusa, B.; Fuh, B.; King, A.; Meier, E.R.; Casella, J.; et al. Health-related quality of life in children with sickle cell anemia: Impact of blood transfusion therapy. Am. J. Hematol. 2015, 90, 139–143. [Google Scholar] [CrossRef]
- Bhatia, M.; Kolva, E.; Cimini, L.; Jin, Z.; Satwani, P.; Savone, M.; George, D.; Garvin, J.; Paz, M.L.; Briamonte, C.; et al. Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2015, 21, 666–672. [Google Scholar] [CrossRef]
- Krishnamurti, L.; Neuberg, D.S.; Sullivan, K.M.; Kamani, N.R.; Abraham, A.; Campigotto, F.; Zhang, W.; Dahdoul, T.; De Castro, L.; Parikh, S.; et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: Results of a prospective multicenter pilot study. Am. J. Hematol. 2019, 94, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, C.; Kamdem, A.; Coïc, L.; Médejel, N.; Lesprit, E.; Hau, I.; Madhi, F.; Lemerle, S.; Biscardi, S.; Bernaudin, F. Acute splenic sequestration in a newborn cohort with sickle cell anemia (SCA): Predictive factors and impact on disease severity. Blood 2010, 116, 263. [Google Scholar]
- Kimaro, F.D.; Jumanne, S.; Sindato, E.M.; Kayange, N.; Chami, N. Prevalence and factors associated with renal dysfunction among children with sickle cell disease attending the sickle cell disease clinic at a tertiary hospital in Northwestern Tanzania. PLoS ONE 2019, 14, e0218024. [Google Scholar] [CrossRef]
- Nouraie, M.; Lee, J.S.; Zhang, Y.; Kanias, T.; Zhao, X.; Xiong, Z.; Oriss, T.B.; Zeng, Q.; Kato, G.J.; Gibbs, J.S.; et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013, 98, 464–472. [Google Scholar] [CrossRef]
- Adams, R.J.; Kutlar, A.; McKie, V.; Carl, E.; Nichols, F.T.; Liu, J.C.; McKie, K.; Clary, A. Alpha thalassemia and stroke risk in sickle cell anemia. Am. J. Hematol. 1994, 45, 279–282. [Google Scholar] [CrossRef]
- Renoux, C.; Connes, P.; Nader, E.; Skinner, S.; Faes, C.; Petras, M.; Bertrand, Y.; Garnier, N.; Cuzzubbo, D.; Divialle-Doumdo, L.; et al. Alpha-thalassaemia promotes frequent vaso-occlusive crises in children with sickle cell anaemia through haemorheological changes. Pediatric Blood Cancer 2017, 64, e26455. [Google Scholar] [CrossRef]
- Bernaudin, F.; Pondarré, C.; Galambrun, C.; Thuret, I. Allogeneic/Matched Related Transplantation for β-Thalassemia and Sickle Cell Anemia. Adv. Exp. Med. Biol. 2017, 1013, 89–122. [Google Scholar]
- Kauf, T.L.; Coates, T.D.; Huazhi, L.; Mody-Patel, N.; Hartzema, A.G. The cost of health care for children and adults with sickle cell disease. Am. J. Hematol. 2009, 84, 323–327. [Google Scholar] [CrossRef]
- Bazuaye, N.; Nwogoh, B.; Ikponmwen, D.; Irowa, O.; Okugbo, S.; Isa, I.; Ighodaro, E.; Aina, Y.I.; Osaguona, A.; Idemudia, O.; et al. First successful allogeneic hematopoietic stem cell transplantation for a sickle cell disease patient in a low resource country (Nigeria): A case report. Ann. Transpl. 2014, 19, 210–213. [Google Scholar]
- Walters, M.C.; Sullivan, K.M.; Bernaudin, F.; Souillet, G.; Vannier, J.P.; Johnson, F.L.; Powars, D.; Bunin, N.; Ohene-Frempong, K. Ohene-Frempong, K. Neurologic complications after allogeneic marrow transplantation for sickle cell anemia. Blood 1995, 85, 879–884. [Google Scholar]
- Gaziev, J.; Marziali, S.; Paciaroni, K.; Isgrò, A.; Marziali, M.; Romigi, A.; Izzi, F.; Mercuri, N.B.; Floris, R.; Palmieri, M.G.; et al. Posterior Reversible Encephalopathy Syndrome after Hematopoietic Cell Transplantation in Children with Hemoglobinopathies. Biol. Blood Marrow Transplant. 2017, 23, 1531–1540. [Google Scholar] [CrossRef] [Green Version]
- Powell, J.D.; Lerner, C.G.; Schwartz, R.H. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J. Immunol. 1999, 162, 2775–2784. [Google Scholar]
- Bolanos1: Bolaños-Meade, J.; Cooke, K.R.; Gamper, C.J.; Ali, S.A.; Ambinder, R.F.; Borrello, I.M.; Fuchs, E.J.; Gladstone, D.E.; Gocke, C.B.; Huff, C.A.; et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: A prospective clinical trial. Lancet Haematol. 2019, 6, e183–e193. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Ribeil, J.A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Tisdale, J.F.; Kanter, J.; Mapara, M.Y.; Kwiatkowski, J.L.; Krishnamurti, L.; Schmidt, M.; Miller, A.L.; Pierciey, F.J.; Shi, W.; Ribeil, J.A.; et al. Current Results of Lentiglobin Gene Therapy in Patients with Severe Sickle Cell Disease Treated Under a Refined Protocol in the Phase 1 Hgb-206 Study. Blood 2018, 132, 1026. [Google Scholar]
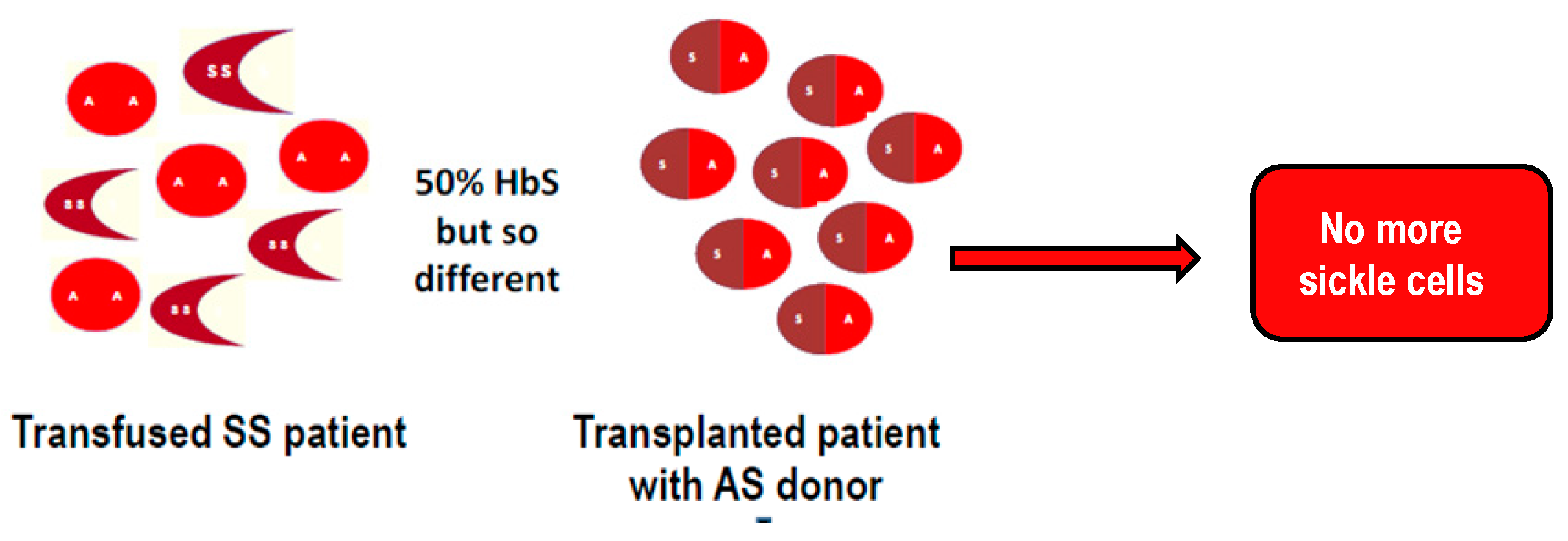


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernaudin, F. Why, Who, When, and How? Rationale for Considering Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease. J. Clin. Med. 2019, 8, 1523. https://doi.org/10.3390/jcm8101523
Bernaudin F. Why, Who, When, and How? Rationale for Considering Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease. Journal of Clinical Medicine. 2019; 8(10):1523. https://doi.org/10.3390/jcm8101523
Chicago/Turabian StyleBernaudin, Françoise. 2019. "Why, Who, When, and How? Rationale for Considering Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease" Journal of Clinical Medicine 8, no. 10: 1523. https://doi.org/10.3390/jcm8101523
APA StyleBernaudin, F. (2019). Why, Who, When, and How? Rationale for Considering Allogeneic Stem Cell Transplantation in Children with Sickle Cell Disease. Journal of Clinical Medicine, 8(10), 1523. https://doi.org/10.3390/jcm8101523