Pathogenetical and Neurophysiological Features of Patients with Autism Spectrum Disorder: Phenomena and Diagnoses


Abstract
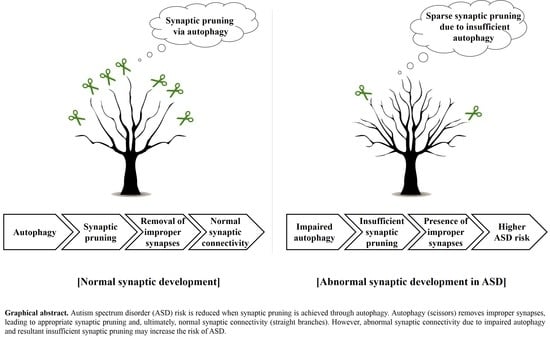
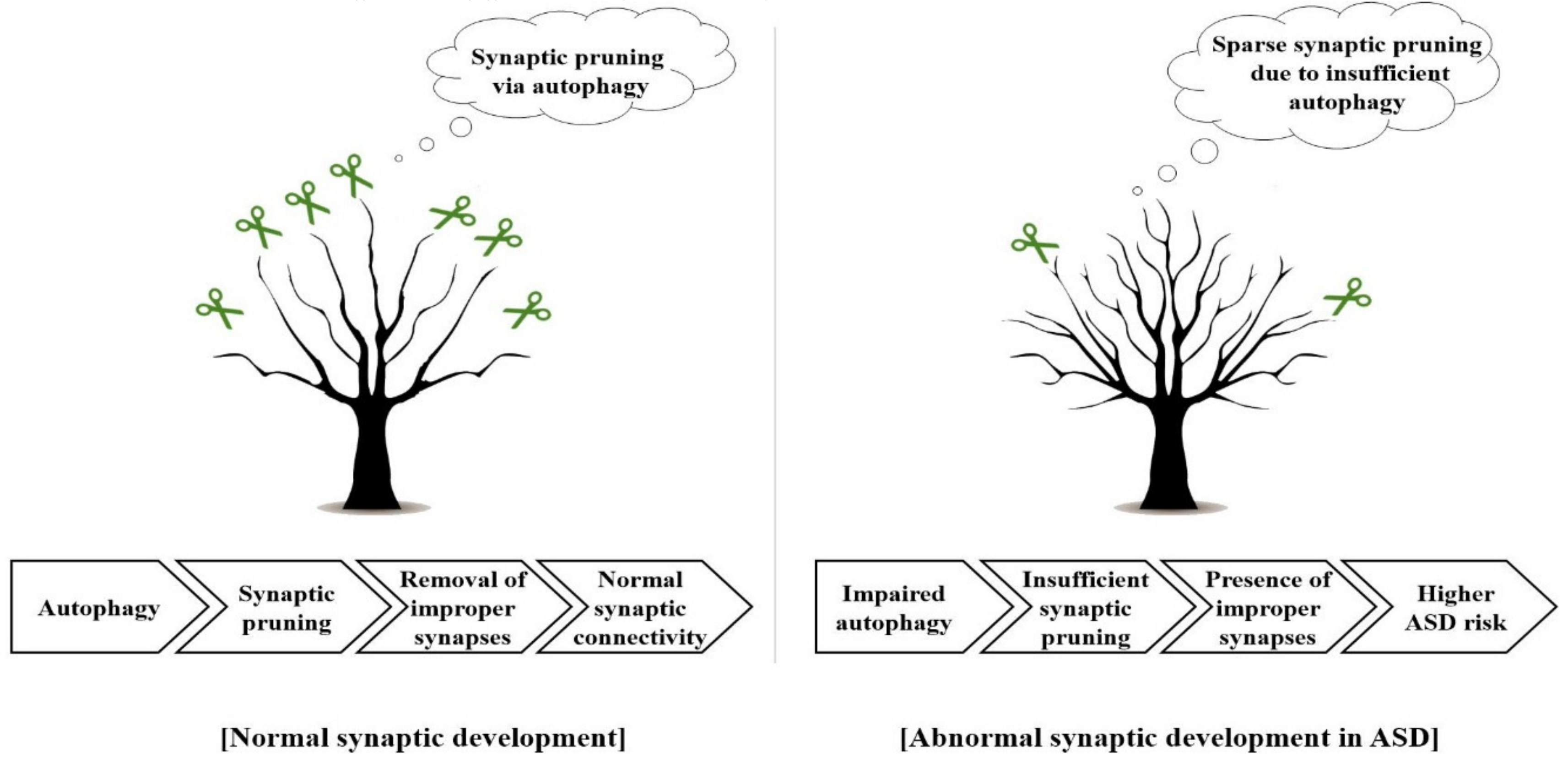
:
1. Introduction
2. The Importance of Synaptic Pruning by Autophagy in ASD Pathophysiology
3. Regulation of Neuroinflammation Might Be Required for the Alleviation of ASD
4. Electrophysiology in ASD
4.1. Detection of Paroxysmal or Epileptic Forms in Resting EEG
4.2. Spatio-Temporal Characteristics of Resting EEG in ASD
4.3. Quantitative Analysis of ERP in ASD
5. Potential Therapeutic Intervention for ASD and Neuropsychiatric Comorbidities
5.1. Melatonin Contributes to Neural Development via Regulating Programmed Cell Death
5.2. Melatonin-Mediated Promotion of Normal Sleep Ameliorates ASD by Facilitating Normal Neural Development
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; BMC Medicine: Arlington, VA, USA, 2013. [Google Scholar]
- Baird, G.; Simonoff, E.; Pickles, A.; Chandler, S.; Loucas, T.; Meldrum, D.; Charman, T. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: The Special Needs and Autism Project (SNAP). Lancet 2006, 368, 210–215. [Google Scholar] [CrossRef]
- Tromans, S.; Chester, V.; Kiani, R.; Alexander, R.; Brugha, T. The prevalence of autism spectrum disorders in adult psychiatric inpatients: A systematic review. Clin. Pract. Epidemiol. Ment. Health 2018, 14, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Yoshimura, I. Prevalence of maladaptive behavior in retarded children as a function of IQ and age. J. Abnorm. Child Psychol. 1978, 6, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Simonoff, E.; Pickles, A.; Charman, T.; Chandler, S.; Loucas, T.; Baird, G. Psychiatric disorders in children with autism spectrum disorders: Prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry 2008, 47, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Osokine, I.; Erlebacher, A. Inflammation and Autism: From Maternal Gut to Fetal Brain. Trends Mol. Med. 2017, 23, 1070–1071. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Maternal infection and immune involvement in autism. Trends Mol. Med. 2011, 17, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Van Erp, T.G.; Hibar, D.P.; Rasmussen, J.M.; Glahn, D.C.; Pearlson, G.D.; Andreassen, O.A.; Agartz, I.; Westlye, L.T.; Haukvik, U.K.; Dale, A.M.; et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol. Psychiatry 2016, 21, 585. [Google Scholar] [CrossRef]
- Satterthwaite, T.D.; Wolf, D.H.; Calkins, M.E.; Vandekar, S.N.; Erus, G.; Ruparel, K.; Roalf, D.R.; Linn, K.A.; Elliott, M.A.; Moore, T.M.; et al. Structural brain abnormalities in youth with psychosis spectrum symptoms. JAMA Psychiatry 2016, 73, 515–524. [Google Scholar] [CrossRef]
- Kanner, L. Autistic disturbances of affective contact. Nervous Child 1943, 2, 217–250. [Google Scholar]
- O’Reilly, C.; Lewis, J.D.; Elsabbagh, M. Is functional brain connectivity atypical in autism? A systemic review of EEG and MEG studies. PLoS ONE 2017, 12, e0175870. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, S.; van Dinteren, R.; Arns, M. Personalized medicine: Review and perspectives of promising baseline EEG biomarkers in major depressive disorder and attention deficit hyperactivity disorder. Neuropsychobiology 2015, 72, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An overview of autism spectrum disorder, heterogeneity and treatment options. Neurosci. Bull. 2017, 33, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.M.; Scahill, L.; McCracken, J.T.; McDougle, C.J.; Aman, M.G.; Tierney, E.; Arnold, L.E.; Martin, A.; Katsovich, L.; Posey, D.J. Effects of short-and long-term risperidone treatment on prolactin levels in children with autism. Biol. Psychiatry 2007, 61, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Robb, A.S.; Andersson, C.; Bellocchio, E.E.; Manos, G.; Rojas-Fernandez, C.; Mathew, S.; Marcus, R.; Owen, R.; Mankoski, R. Safety and tolerability of aripiprazole in the treatment of irritability associated with autistic disorder in pediatric subjects (6–17 years Old): Results from a pooled analysis of 2 studies. Prim. Care Companion Disord. 2011, 13, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Jin, Y.; Choi, J.; Park, S.; Lee, T.H.; Lee, S.R.; Chang, K.T.; Hong, Y. Melatonin as a novel interventional candidate for Fragile X syndrome with autism spectrum disorder in humans. Int. J. Mol. Sci. 2017, 18, 1314. [Google Scholar] [CrossRef]
- Wong, K.; Leonard, H.; Jacoby, P.; Ellaway, C.; Downs, J. The trajectories of sleep disturbance in Rett syndrome. J. Sleep Res. 2015, 24, 223–233. [Google Scholar] [CrossRef]
- Hancock, E.; O’Callaghan, F.; Osborne, J.P. Effect of melatonin dosage on sleep disorder in tuberous sclerosis complex. J. Child Neurol. 2005, 20, 78–80. [Google Scholar] [CrossRef]
- Park, G.; Lee, S.H.; Oh, D.S.; Kim, Y.U. Melatonin inhibits neuronal dysfunction-associated with neuroinflammation by atopic psychological stress in NC/Nga atopic-like mouse models. J. Pineal Res. 2017, 63, e12420. [Google Scholar] [CrossRef]
- Permpoonputtana, K.; Tangweerasing, P.; Mukda, S.; Boontem, P.; Nopparat, C.; Govitrapong, P. Long-term administration of melatonin attenuates neuroinflammation in the aged mouse brain. EXCLI J. 2018, 17, 634–646. [Google Scholar]
- Hutsler, J.J.; Zhang, H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brian Res. 2010, 1309, 83–94. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopemtal disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [PubMed]
- Navlakha, S.; Bar-Joseph, Z.; Barth, A.L. Network design and the brain. Trends Cogn. Sci. 2018, 22, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.G.; Hawksworth, O.A.; Woodruff, T.M. Complement: The emerging architect of the developing brain. Trends Neurosci. 2018, 41, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Lieberman, O.J.; McGuirt, A.F.; Tang, G.; Sulzer, D. Roles for neuronal and glial autophagy in synaptic pruning during development. Neurobiol Dis. 2019, 122, 49–63. [Google Scholar] [CrossRef]
- Keshavan, M.S.; Anderson, S.; Pettegrew, J.W. Is schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J. Psychiatr. Res. 1994, 28, 239–265. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Hong, Y.; Park, C.Y.; Hong, Y. Molecular interactions of autophagy with the immune system and cancer. Int. J. Mol. Sci. 2017, 18, 1694. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Axonal autophagy: Mini-review for autophagy in the CNS. Neurosci. Lett. 2019, 697, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; Li, Y.; Baker, S.J.; Parada, L.F. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef]
- Chana, G.; Laskaris, L.; Pantelis, C.; Gillett, P.; Testa, R.; Zantomio, D.; Burrows, E.L.; Hannan, A.J.; Everall, I.P.; Skafidas, E. Decreased expression of mGluR5 within the dorsolateral prefrontal cortex in autism and increased microglial number in mGluR5 knockout mice: Pathophysiological and neurobehavioral implications. Brain Behav. Immun. 2015, 49, 197–205. [Google Scholar] [CrossRef]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, U.; Murray, P.J.; Urwyler, A.; Yee, B.K.; Schedlowski, M.; Feldon, J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol. Psychiatry 2008, 13, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Won, E. The influence of stress on neuroinflammation and alterations in brain structure and function in major depressive disorder. Behav. Brain Res. 2017, 329, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Frodl, T.; Carballedo, A.; Hughes, M.M.; Saleh, K.; Fagan, A.; Skokauskas, N.; McLoughlin, D.M.; Meaney, J.; O’Keane, V.; Connor, T.J. Reduced expression of glucocorticoid-inducible genes GILZ and SGK-1: High IL-6 levels are associated with reduced hippocampal volumes in major depressive disorder. Transl. Psychiatry 2012, 2, e88. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, S.; Won, J.; Jin, Y.; Hong, Y.; Hur, T.Y.; Kim, J.H.; Lee, S.R.; Hong, Y. Pathophysiological and neurobehavioral characteristics of a propionic acid-mediated autism-like rat model. PLoS ONE 2018, 13, e0192925. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Faluyi, Y.O.; Hong, Y.T.; Fryer, T.D.; Mak, E.; Gabel, S.; Hayes, L.; Soteriades, S.; Williams, G.B.; Arnold, R.; et al. Neuroinflammatory and morphological changes in late-life depression: The NIMROD study. Br. J. Psychiatry 2016, 209, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-induced anhedonia: Endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef]
- Kreisel, T.; Frank, M.G.; Licht, T.; Reshef, R.; Ben-Menachem-Zidon, O.; Baratta, M.V.; Maier, S.F.; Yirmiya, R. Dynamic microglia alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 2014, 19, 699–709. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Stewart, J.M.; Panagiotidou, S.; Melamed, I. Mast cell, brain inflammation and autism. Eur. J. Pharmacol. 2016, 778, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Abramson, I.; Semendeferi, K.; Courchesne, E.; Everall, I.P. Abnormal microglia-neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res. 2012, 1456, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef] [PubMed]
- Le Belle, J.E.; Sperry, J.; Ngo, A.; Ghochani, Y.; Laks, D.R.; López-Aranda, M.; Silva, A.J.; Kornblum, H.I. Maternal inflammation contributes to brain overgrowth and autism-associated behaviors through altered redox signaling in stem and progenitor cells. Stem Cell Rep. 2014, 3, 725–734. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglia activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Thomas, T.C.; Ziebell, J.M.; Pauly, J.R.; Lifshitz, J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 2012, 225, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, K.; Wang, H.; Xu, J.; Lu, X.; Zhang, L.; Zhu, L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: Possible involvement of mTOR pathway. Neurochem. Int. 2014, 76, 23–31. [Google Scholar] [CrossRef]
- Noachtar, S.; Rémi, J. The role of EEG in epilepsy: A critical review. Epilepsy Behav. 2009, 15, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, A.; Loo, S.K. Use of EEG to diagnose ADHD. Curr. Psychiatry Rep. 2014, 16, 498. [Google Scholar] [CrossRef]
- De Aguiar Neto, F.S.; Rosa, J.L.G. Depression biomarkers using non-invasive EEG: A review. Neurosci. Biobehav. Rev. 2019, 105, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barstein, J.; Ethridge, L.E.; Mosconi, M.W.; Takarae, Y.; Sweeney, J.A. Resting state EEG abnormalities in autism spectrum disorders. J. Neurodev. Disord. 2013, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Boutros, N.N.; Lajiness-ÓNeill, R.; Zillgitt, A.; Richard, A.E.; Bowyer, S.M. EEG changes associated with autistic spectrum disorders. Neuropsychiatr. Electrophysiol. 2015, 1, 1–20. [Google Scholar] [CrossRef]
- Jeste, S.S.; Nelson, C.A., 3rd. Event related potentials in the understanding of autism spectrum disorders: An analytical review. J. Autism Dev. Disord. 2009, 39, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Marco, E.J.; Hinkley, L.B.; Hill, S.S.; Nagarajan, S.S. Sensory processing in autism: A review of neurophysiologic findings. Pediatr. Res. 2011, 69, 48R–54R. [Google Scholar] [CrossRef] [PubMed]
- Modi, M.E.; Sahin, M. Translational use of event-related potentials to assess circuit integrity in ASD. Nat. Rev. Neurol. 2017, 13, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Spence, S.J.; Schneider, M.T. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr. Res. 2009, 65, 599–606. [Google Scholar] [CrossRef]
- Strasser, L.; Downes, M.; Kung, J.; Cross, J.H.; De Haan, M. Prevalence and risk factors for autism spectrum disorder in epilepsy: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2018, 60, 19–29. [Google Scholar] [CrossRef]
- Sánchez Fernández, I.; Loddenkemper, T.; Peters, J.M.; Kothare, S.V. Electrical status epilepticus in sleep: Clinical presentation and pathophysiology. Pediatr. Neurol. 2012, 47, 390–410. [Google Scholar] [CrossRef]
- Kagan-Kushnir, T.; Roberts, S.W.; Snead, O.C. 3rd. Screening electroencephalograms in autism spectrum disorders: Evidence-based guideline. J. Child Neurol. 2005, 20, 197–206. [Google Scholar] [CrossRef]
- Frye, R.E.; Butler, I.; Strickland, D.; Castillo, E.; Papanicolaou, A. Electroencephalogram discharges in atypical cognitive development. J. Child Neurol. 2010, 25, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Khoo, H.M.; von Ellenrieder, N.; Zazubovits, N.; Gotman, J. DeepIED: An epileptic discharge detector for EEG=fMRI based on deep learning. Neuroimage Clin. 2017, 17, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Hussein, R.; Palangi, H.; Ward, R.K.; Wang, Z.J. Optimized deep neural network architecture for robust detection of epileptic seizures using EEG signals. Clin. Neurophysiol. 2019, 130, 25–37. [Google Scholar] [CrossRef] [PubMed]
- A long short-term memory deep learning network for the prediction of epileptic seizures using EEG signals. Comput. Biol. Med. 2018, 99, 24–37. [CrossRef] [PubMed]
- Tjepkema-Cloostermans, M.C.; de Carvalho, R.C.V.; van Putten, M.J.A.M. Deep learning for detection of focal epileptiform discharges from scalp EEG recordings. Clin. Neurophysiol. 2018, 129, 2191–2196. [Google Scholar] [CrossRef] [PubMed]
- Pérez Velázquez, J.L.; Galán, R.F. Information gain in the brain’s resting state: A new perspective on autism. Front. Neuroinform. 2013, 7, 37. [Google Scholar] [CrossRef]
- Murias, M.; Webb, S.J.; Greenson, J.; Dawson, G. Resting state cortical connectivity reflected in EEG coherence in individuals with autism. Biol. Psychiatry 2007, 62, 270–273. [Google Scholar] [CrossRef]
- Van Diessen, E.; Senders, J.; Jansen, F.E.; Boersma, M.; Bruining, H. Increased power of resting-state gamma oscillations in autism spectrum disorder detected by routine electroencephalography. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 537–540. [Google Scholar] [CrossRef]
- Tierney, A.L.; Gabard-Durnam, L.; Vogel-Farley, V.; Tager-Flusberg, H.; Nelson, C.A. Developmental trajectories of resting EEG power: An endophenotype of autism spectrum disorder. PLoS ONE 2012, 7, e39127. [Google Scholar] [CrossRef]
- Ahmadlou, M.; Adeli, H.; Adeli, A. Fractality and a wavelet-chaos-neural network methodology for EEG-based diagnosis of autistic spectrum disorder. J. Clin. Neurophysiol. 2010, 27, 328–333. [Google Scholar] [CrossRef]
- Kouijzer, M.E.; van Schie, H.T.; Gerrits, B.J.; Buitelaar, J.K.; de Moor, J.M. Is EEG-biofeedback an effective treatment in autism spectrum disorders? A randomized controlled trial. Appl. Psychophysiol. Biofeedback 2013, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Pineda, J.A.; Carrasco, K.; Datko, M.; Pillen, S.; Schalles, M. Neurofeedback training produces normalization in behavioural and electrophysiological measures of high-functioning autism. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurt, E.; Arnold, L.E.; Lofthouse, N. Quantitative EEG neurofeedback for the treatment of pediatric attention-deficit/hyperactivity disorder, autism spectrum disorders, learning disorders, and epilepsy. Child Adolesc. Psychiatr. Clin. N. Am. 2014, 23, 465–486. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Webb, S.J.; Wijsman, E.; Schellenberg, G.; Estes, A.; Munson, J.; Faja, S. Neurocognitive and electrophysiological evidence of altered face processing in parents of children with autism: Implications for a model of abnormal development of social brain circuitry in autism. Dev. Psychopathol. 2005, 17, 679–697. [Google Scholar] [CrossRef]
- Takarae, Y.; Luna, B.; Minshew, N.J.; Sweeney, J.A. Visual motion processing and visual sensorimotor control in autism. J. Int. Neuropsychol. Soc. 2014, 20, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Matic, V.; Cherian, P.J.; Koolen, N.; Ansari, A.H.; Naulaers, G.; Govaert, P.; Van Huffel, S.; De Vos, M.; Vanhatalo, S. Objective differentiation of neonatal EEG background grades using detrended fluctuation analysis. Front. Hum. Neurosci. 2015, 9, 189. [Google Scholar] [CrossRef]
- Lee, J.M.; Kim, D.J.; Kim, I.Y.; Suk Park, K.; Kim, S.I. Nonlinear-analysis of human sleep EEG using detrended fluctuation analysis. Med. Eng. Phys. 2004, 26, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Croce, P.; Quercia, A.; Costa, S.; Zappasodi, F. Circadian rhythms in fractal features of EEG signals. Front. Physiol. 2018, 9, 1567. [Google Scholar] [CrossRef]
- Wilson, T.W.; Rojas, D.C.; Reite, M.L.; Teale, P.D.; Rogers, S.J. Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biol. Psychiatry 2007, 62, 192–197. [Google Scholar] [CrossRef]
- Rojas, D.C.; Bawn, S.D.; Benkers, T.L.; Reite, M.L.; Rogers, S.J. Smaller left hemisphere planum temporale in adults with autistic disorder. Neurosci. Lett. 2002, 328, 237–240. [Google Scholar] [CrossRef]
- Vissers, M.E.; Cohen, M.X.; Geurts, H.M. Brain connectivity and high functioning autism: A promising path of research that needs refined models, methodological convergence, and stronger behavioral links. Neurosci. Biobehav. Rev. 2012, 36, 604–625. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Pierce, K. Why the frontal cortex in autism might be talking only to itself: Local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol. 2005, 15, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Maximo, J.O.; Keown, C.L.; Nair, A.; Müller, R.A. Approaches to local connectivity in autism using resting state functional connectivity MRI. Front. Hum. Neurosci. 2013, 7, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starck, T.; Nikkinen, J.; Rahko, J.; Remes, J.; Hurtig, T.; Haapsamo, H.; Jussila, K.; Kuusikko-Gauffin, S.; Mattila, M.L.; Jansson-Verkasalo, E.; et al. Resting state fMRI reveals a default mode dissociation between retrosplenial and medial prefrontal subnetworks in ASD despite motion scrubbing. Front. Hum. Neurosci. 2013, 7, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosl, W.; Tierney, A.; Tager-Flusberg, H.; Nelson, C. EEG complexity as a biomarker for autism spectrum disorder risk. BMC Med. 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, R.W.; North, D.M.; Biver, C.J. Self-organized criticality and the development of EEG phase reset. Hum. Brain Mapp. 2009, 30, 553–574. [Google Scholar] [CrossRef]
- Wang, S.; Yang, C.; Liu, Y.; Shao, Z.; Jackson, T. Early and late stage processing abnormalities in autism spectrum disorders: An ERP study. PLoS ONE 2017, 12, e0178542. [Google Scholar] [CrossRef]
- Chien, Y.L.; Hsieh, M.H.; Gau, S.S. P50-N100-P200 sensory gating deficits in adolescents and young adults with autism spectrum disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 95, 109683. [Google Scholar] [CrossRef]
- Chaby, L.; George, N.; Renault, B.; Fiori, N. Age-related changes in brain responses to personally known faces: An event-related potential (ERP) study in humans. Neurosci. Lett. 2003, 349, 125–129. [Google Scholar] [CrossRef]
- Cui, T.; Wang, P.P.; Liu, S.; Zhang, X. P300 amplitude and latency in autism spectrum disorder: A meta-analysis. Eur. Child Adolesc. Psychiatry 2017, 26, 177–190. [Google Scholar] [CrossRef]
- De Haan, M.; Johnson, M.H.; Halit, H. Development of face-sensitive event-related potentials during infancy: A review. Int. J. Psychophysiol. 2003, 51, 45–58. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Jiang, Y.; Yin, Z.; Jiang, K.; Li, L.; Shuai, J. Optimal pathways for the assembly of the Apaf-1· cytochrome c complex into apoptosome. Phys. Chem. Chem. Phys. 2018, 20, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, F.; Alvarez-Bolado, G.; Meyer, B.I.; Roth, K.A.; Gruss, P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998, 94, 727–737. [Google Scholar] [CrossRef]
- Motoyama, N.; Wang, E.; Roth, K.A.; Sawa, H.; Nakayama, K.; Nakayama, K.; Negishi, I.; Senju, S.; Zhang, Q.; Fujii, S. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995, 267, 1506–1510. [Google Scholar] [CrossRef]
- Cheung, R.T. The utility of melatonin in reducing cerebral damage resulting from ischemia and reperfusion. J. Pineal Res. 2003, 34, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Reina, M.; Martínez, A. A new free radical scavenging cascade involving melatonin and three of its metabolites (3OHM, AFMK and AMK). Comput. Theor. Chem. 2018, 1123, 111–118. [Google Scholar] [CrossRef]
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and regulating pro- and anti-apoptotic protein levels. Neuroscience 2005, 135, 879–886. [Google Scholar] [CrossRef]
- Koh, P.O. Melatonin attenuates the focal cerebral ischemic injury by inhibiting the dissociation of pBad from 14-3-3. J. Pineal Res. 2008, 44, 101–106. [Google Scholar] [CrossRef]
- Heiskanen, K.M.; Bhat, M.B.; Wang, H.W.; Ma, J.; Nieminen, A.L. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J. Biol. Chem. 1999, 274, 5654–5658. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates and apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Guégan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS. Neurosci. Ther. 2009, 15, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Geden, M.J.; Romero, S.E.; Deshmukh, M. Apoptosis versus Axon Pruning: Molecular Intersection of Two Distinct Pathways for Axon Degeneration. Neurosci. Res. 2019, 139, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Moretti, E.; Bonomini, F.; Reiter, R.J.; Rodella, L.F.; Rezzani, R. Promising antineoplastic actions of melatonin. Front. Pharmacol. 2018, 9, 1086. [Google Scholar] [CrossRef]
- Tian, Y.; Yabuki, Y.; Moriguchi, S.; Fukunaga, K.; Mao, P.J.; Hong, L.J.; Lu, Y.M.; Wang, R.; Ahmed, M.M.; Liao, M.H. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J. Pineal Res. 2014, 56, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Sharma, B.M.; Sharma, B. Benefits of agomelatine in behavioral, neurochemical and blood brain barrier alterations in prenatal valproic acid induced autism spectrum disorder. Neurochem. Int. 2015, 91, 34–45. [Google Scholar] [CrossRef]
- Luo, C.; Yang, Q.; Liu, Y.; Zhou, S.; Jiang, J.; Reiter, R.J.; Bhattacharya, P.; Cui, Y.; Yang, H.; Ma, H.; et al. The multiple protective roles and molecular mechanisms of melatonin and its precursor N-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic. Biol. Med. 2018, 130, 215–233. [Google Scholar] [CrossRef]
- Marsh, D.; Dragich, J.M. Autophagy in mammalian neurodevelopment and implications for childhood neurological disorders. Neurosci. Lett. 2019, 697, 29–33. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Koekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmentaldisability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Song, G.; Panoutsopoulos, A.; Riyadh, M.A.; Kaushik, G.; Halmai, J.; Levenson, R.; Zarbalis, K.S.; Giulivi, C. Beyond autophagy: A novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 2018, 8, 11348. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H. Genetics of autism spectrum disorder: Current status and possible clinical applications. Exp. Neurobiol. 2015, 24, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Yamada, K.; Iwayama, Y.; Toyota, T.; Matsuzaki, H.; Miyachi, T.; Yamada, S.; Tsujii, M.; et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol. Autism 2012, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, D.; Lachman, H.M.; Zheng, D. Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl. Psychiatry 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.L.; Pramparo, T.; Winn, M.E.; Barnes, C.C.; Li, H.R.; Weiss, L.; Fan, J.B.; Murray, S.; April, C.; Belinson, H.; et al. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet. 2012, 8, e1002592. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, J.P. II.; Pegoraro, L.F.L.; Lazzeroni, L.C.; Raman, M.M.; Hallmayer, J.F.; Monterrey, J.C.; Cleveland, S.C.; Wolke, O.N.; Phillips, J.M.; Reiss, A.L.; et al. Genetic and environmental influences on structural brain measures in twins with autism spectrum disorder. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Gómez, R.L.; Edgin, J.O. Sleep as a window into early neural development: Shifts in sleep-dependent learning effects across early childhood. Child Dev. Perspect. 2015, 9, 183–189. [Google Scholar] [CrossRef]
- Kurth, S.; Olini, N.; Huber, R.; LeBourgeois, M. Sleep and early cortical development. Curr. Sleep Med. Rep. 2015, 1, 64–73. [Google Scholar] [CrossRef]
- Dumoulin Bridi, M.C.; Aton, S.J.; Seibt, J.; Renouard, L.; Coleman, T.; Frank, M.G. Rapid eye movement sleep promotes cortical plasticity in the developing brain. Sci. Adv. 2015, 1, e1500105. [Google Scholar] [CrossRef] [Green Version]
- Pagani, L.; St Clair, P.A.; Teshiba, T.M.; Service, S.K.; Fears, S.C.; Araya, C.; Araya, X.; Bejarano, J.; Ramirez, M.; Castrillón, G. Genetic contributions to circadian activity rhythm and sleep pattern phenotypes in pedigrees segregating for severe bipolar disorder. Proc. Natl. Acad. Sci. USA 2016, 113, E754–E761. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.A.; Simpson, F.C. Obstructive sleep apnea and psychiatric disorders: A systematic review. J. Clin. Sleep Med. 2015, 11, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.M.; Videnovic, A. Chronic sleep disturbance and neural injury: Links to neurodegenerative disease. Nat. Sci. Sleep 2016, 8, 55–61. [Google Scholar] [PubMed]
- Morris, G.; Stubbs, B.; Köhler, C.A.; Walder, K.; Slyepchenko, A.; Berk, M.; Carvalho, A.F. The putative role of oxidative stress and inflammation in the pathophysiology of sleep dysfunction across neuropsychiatric disorders: Focus on chronic fatigue syndrome, bipolar disorder and multiple sclerosis. Sleep Med. Rev. 2018, 41, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Vázquez, J.; Camacho-Arroyo, I.; Velázquez-Moctezuma, J. Differential impact of REM sleep deprivation on cytoskeletal proteins of brain regions involved in sleep regulation. Neuropsychobiology 2012, 65, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Grosmark, A.D.; Mizuseki, K.; Pastalkova, E.; Diba, K.; Buzsáki, G. REM sleep reorganizes hippocampal excitability. Neuron 2012, 75, 1001–1007. [Google Scholar] [CrossRef]
- Davis, C.J.; Zielinski, M.R.; Dunbrasky, D.; Taishi, P.; Dinarello, C.A.; Krueger, J.M. Interleukin 37 expression in mice alters sleep responses to inflammatory agents and influenza virus infection. Neurobiol. Sleep Circadian Rhythms 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.M. The role of cytokines in sleep regulation. Curr. Pharm. Des. 2008, 14, 3408–3416. [Google Scholar] [CrossRef] [PubMed]
- Mieda, M.; Ono, D.; Hasegawa, E.; Okamoto, H.; Honma, K.; Honma, S.; Sakurai, T. Cellular clocks in AVP neurons of the SCN are critical for interneuronal coupling regulating circadian behavior rhythm. Neuron 2015, 85, 1103–1116. [Google Scholar] [CrossRef]
- Waly, N.E.; Hallworth, R. Circadian pattern of melatonin MT1 and MT2 receptor localization in the rat suprachiasmatic nucleus. J. Circadian Rhythms 2015, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.; Reiter, R.J.; Pérez-San-Gregorio, M.A. Melatonin as a potential therapeutic agent in psychiatric illness. Hum. Psychopharmacol. 2009, 24, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Houdek, P.; Polidarova, L.; Novakova, M.; Mateju, K.; Kubik, S.; Sumova, A. Melatonin administered during the fetal stage affects circadian clock in the suprachiasmatic nucleus but not in the liver. Dev. Neurobiol. 2015, 75, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Garzón, C.; Guerrero, J.M.; Aramburu, O.; Guzmán, T. Effect of melatonin administration on sleep, behavioral disorders and hypnotic drug discontinuation in the elderly: A randomized, double-blind, placebo-controlled study. Aging Clin. Exp. Res. 2009, 21, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Kayumov, L.; Brown, G.; Jindal, R.; Buttoo, K.; Shapiro, C.M. A randomized, double-blind, placebo-controlled crossover study of the effects of exogenous melatonin on delayed sleep phase syndrome. Psychosom. Med. 2001, 63, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Veatch, O.J.; Goldman, S.E.; Adkins, K.W.; Malow, B.A. Melatonin in children with autism spectrum disorders: How does the evidence fit together? J. Nat. Sci. 2015, 1, e125. [Google Scholar] [PubMed]
- Rossignol, D.A.; Frye, R.E. Melatonin in autism spectrum disorders. Curr. Clin. Pharmacol. 2014, 9, 326–334. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Dysregulated genes listed | Ref. |
|---|---|---|
| Dorsolateral prefrontal cortex | Reduction in mGluR5 | [41] |
| Anterior cingulate gyrus, motor cortex, thalamus | Reduction in Metaxin 2 (MTX2), Light polypeptide (NEFL), Solute carrier family 25, member 27 (SLC25A27) | [123] |
| Inhibitory neurons in brain | Augmentation in GAD1, RELN, VIP, CHD7, PAX6, TBX1, CHD8, EHMT1, SATB2 | [124] |
| Dorsolateral prefrontal cortex | Dysregulation of ErbB4, MMP2, NID1, TIMP1, COL4A3, RELN, ROBO1, ADORA2A, p21 (CDKN1A), 14- 3-3, HGF, FGFRL1, TSC1 | [125] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Choi, J.; Lee, S.; Kim, J.W.; Hong, Y. Pathogenetical and Neurophysiological Features of Patients with Autism Spectrum Disorder: Phenomena and Diagnoses. J. Clin. Med. 2019, 8, 1588. https://doi.org/10.3390/jcm8101588
Jin Y, Choi J, Lee S, Kim JW, Hong Y. Pathogenetical and Neurophysiological Features of Patients with Autism Spectrum Disorder: Phenomena and Diagnoses. Journal of Clinical Medicine. 2019; 8(10):1588. https://doi.org/10.3390/jcm8101588
Chicago/Turabian StyleJin, Yunho, Jeonghyun Choi, Seunghoon Lee, Jong Won Kim, and Yonggeun Hong. 2019. "Pathogenetical and Neurophysiological Features of Patients with Autism Spectrum Disorder: Phenomena and Diagnoses" Journal of Clinical Medicine 8, no. 10: 1588. https://doi.org/10.3390/jcm8101588