The Clinicopathological Spectrum of Acromegaly



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Primary Pituitary Causes
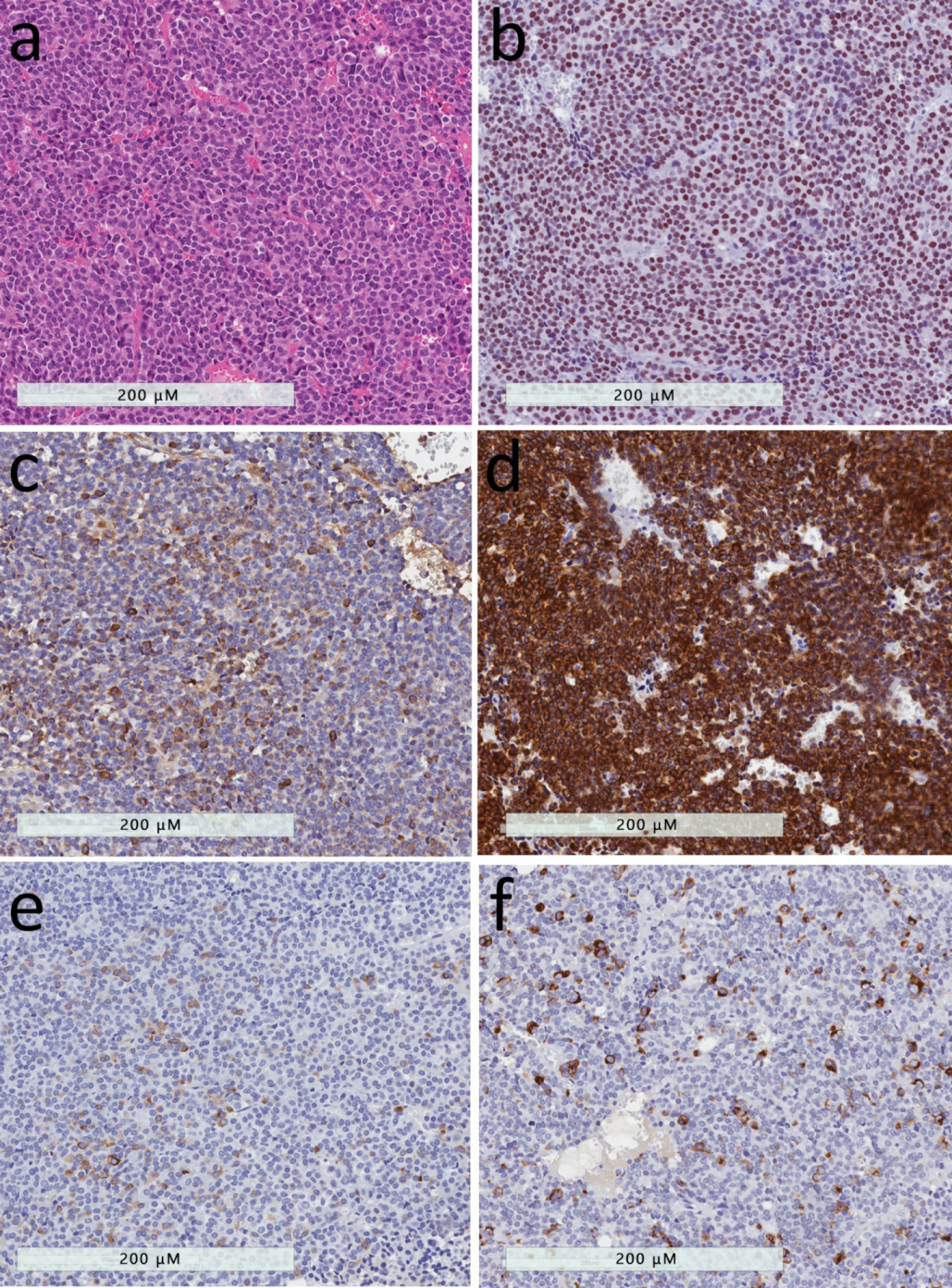
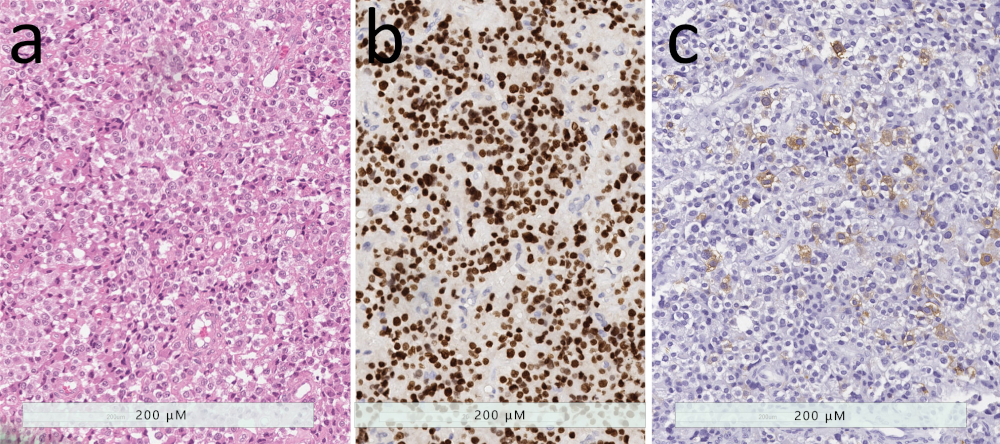
2.1. Densely-granulated Somatotroph Tumor
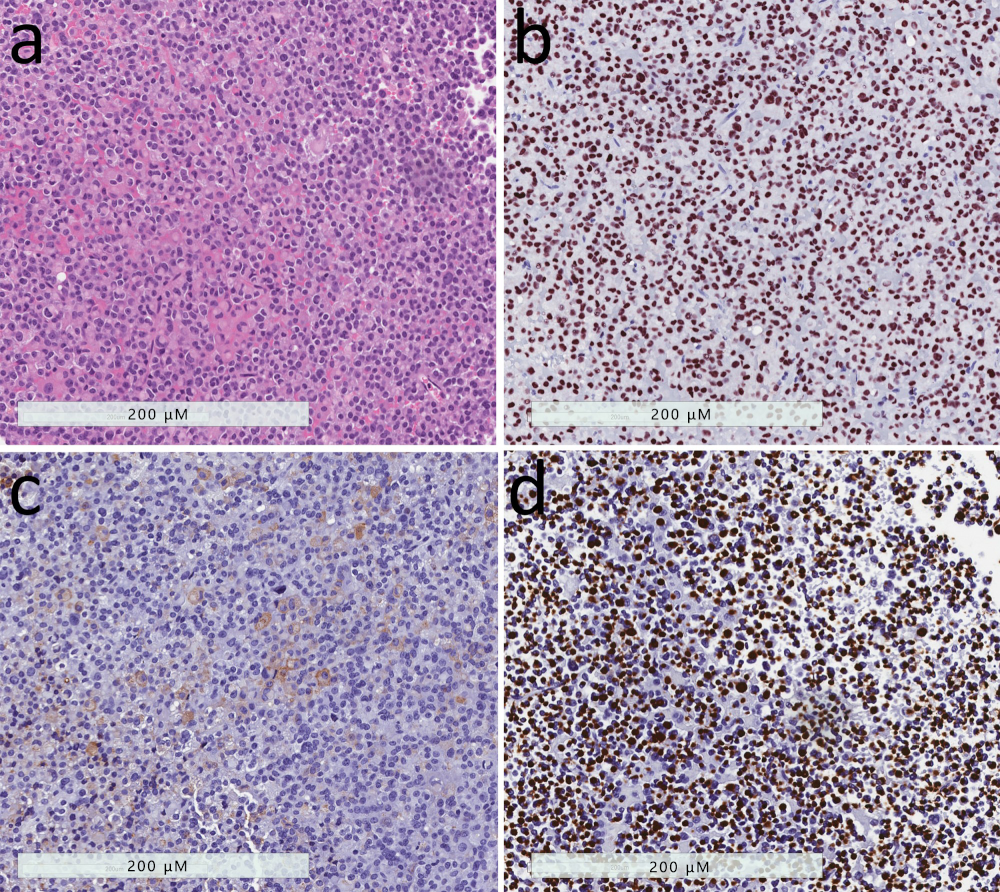
2.2. Sparsely-Granulated Somatotroph Tumor
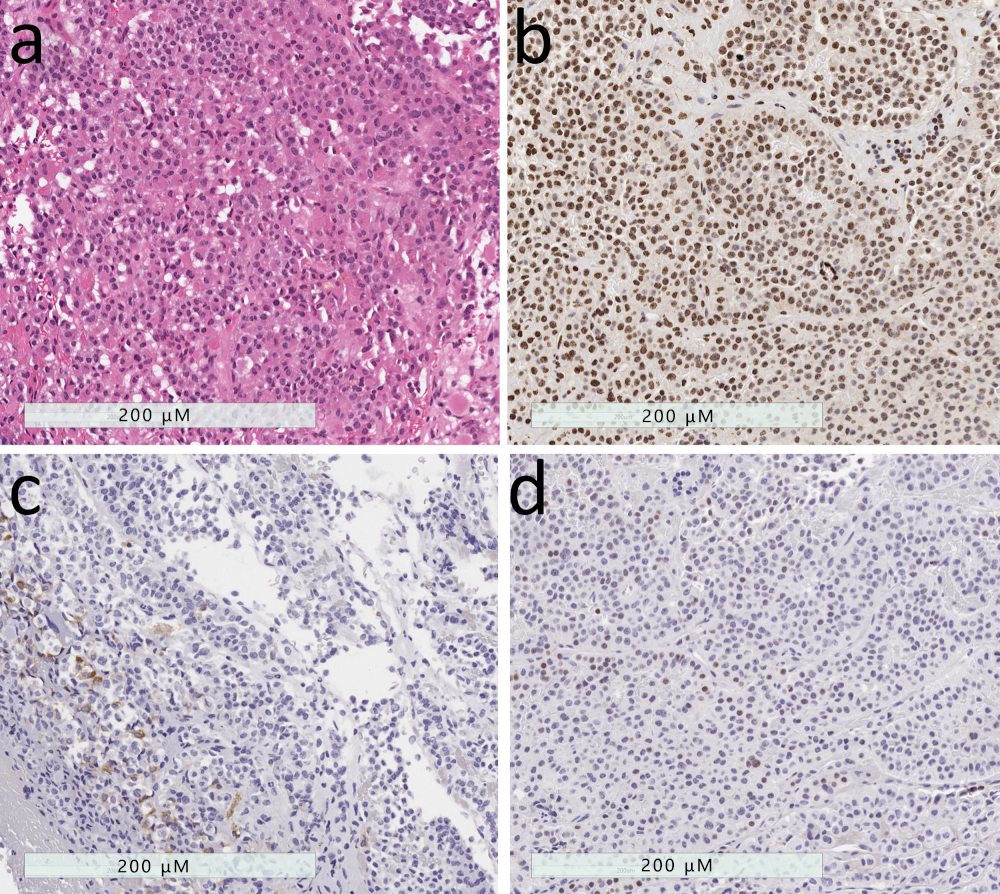
2.3. Mammosomatotroph Tumor
2.4. Mature Plurihormomal Pit1-Lineage Tumor
2.5. Mixed Somatotroph-Lactotroph Tumor
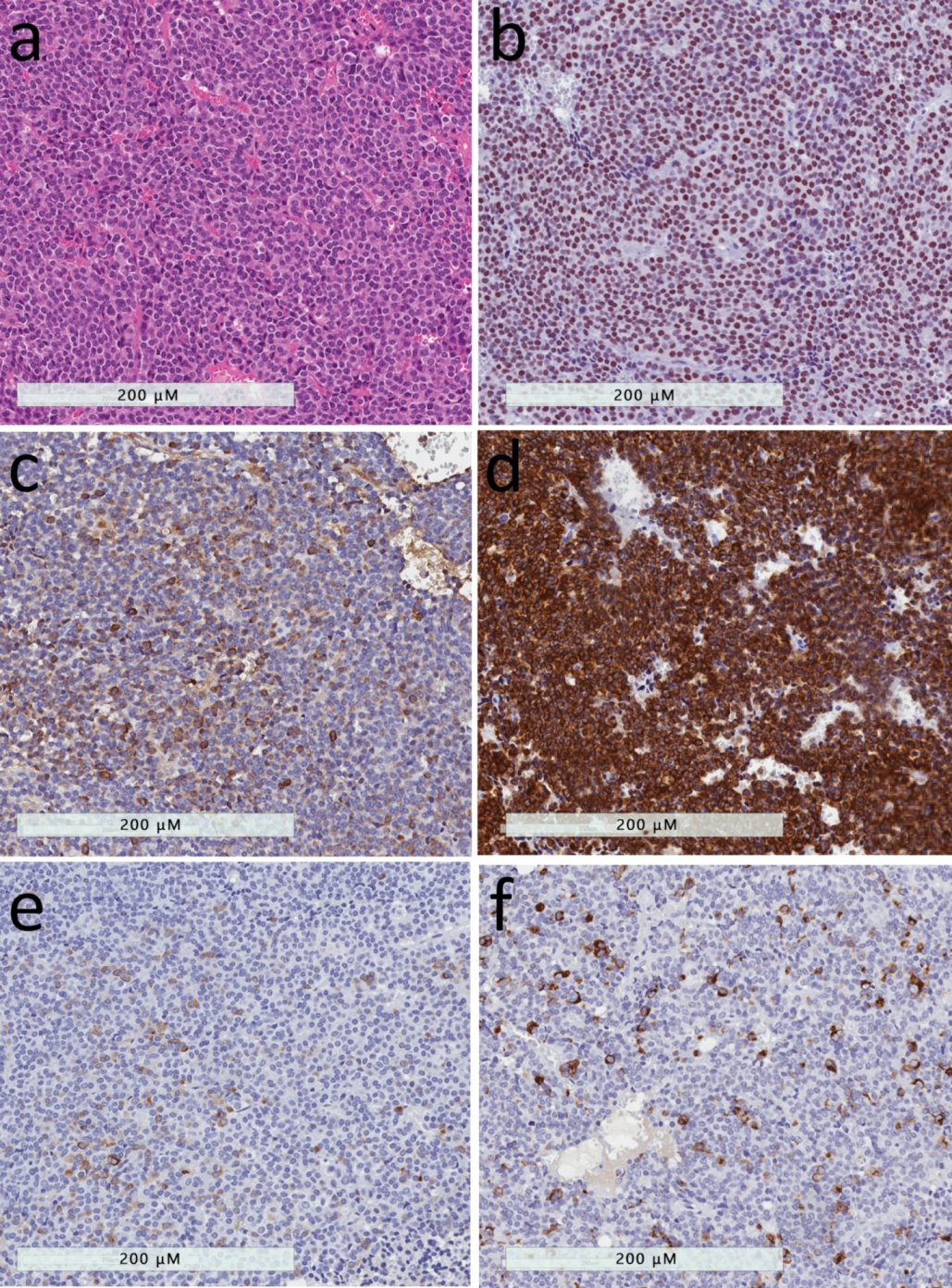
2.6. Acidophil Stem Cell Tumor
2.7. Poorly Differentiated Pit1-Lineage Tumor
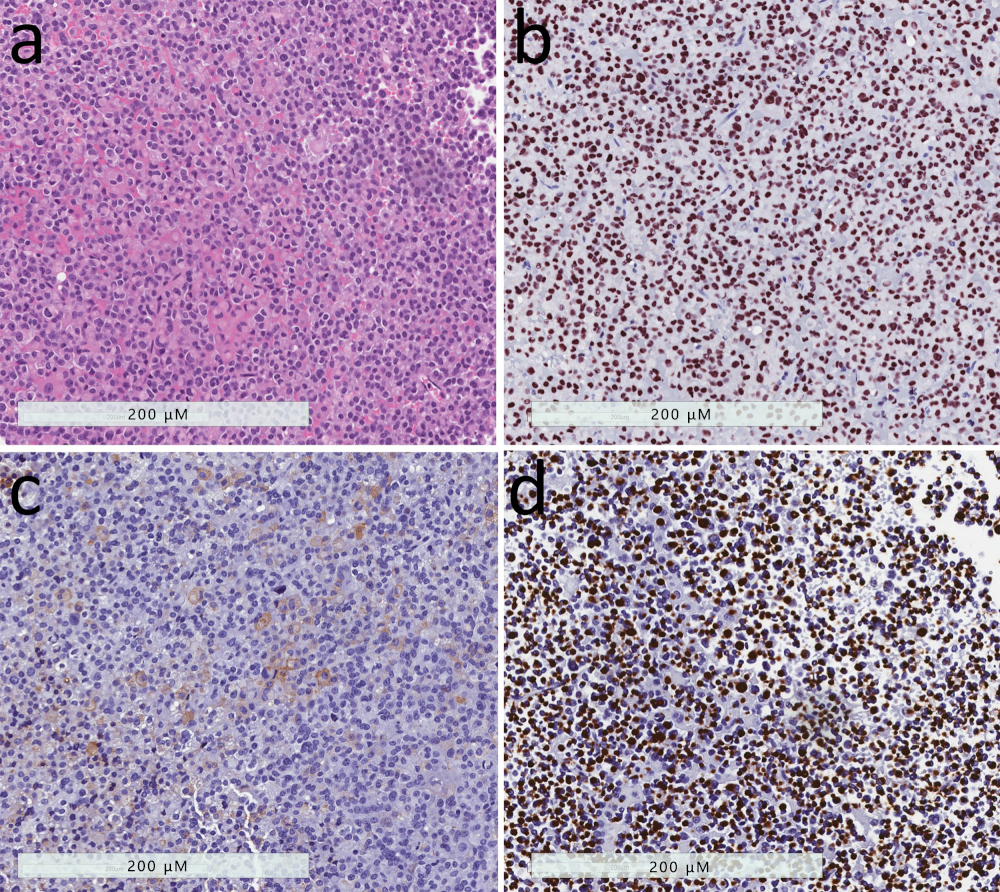
2.8. Pituitary Carcinoma
2.9. Primary Pituitary Hyperplasia
3. Extra-Pituitary Tumors
3.1. Ectopic Growth Hormone Hypersecretion
3.2. Excess Production of Growth Hormone-Releasing Hormone
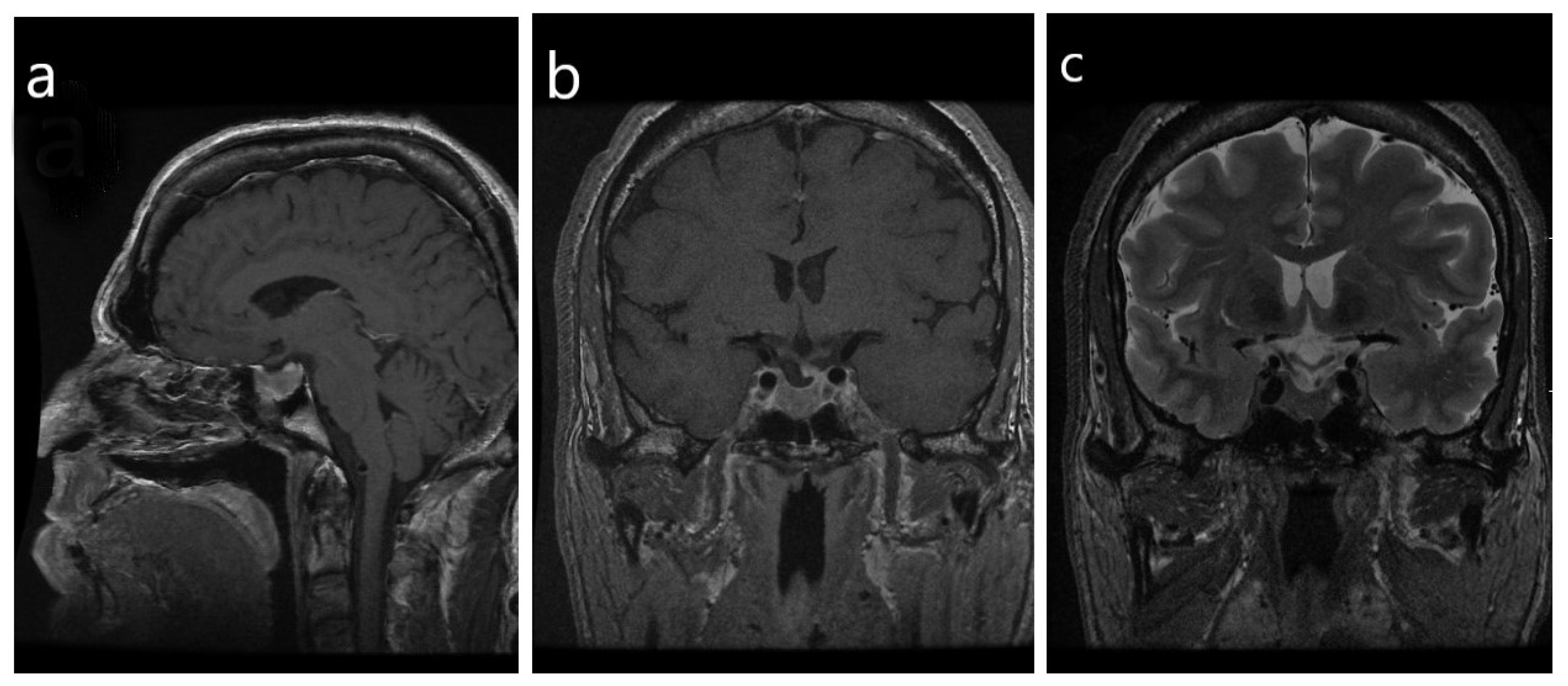
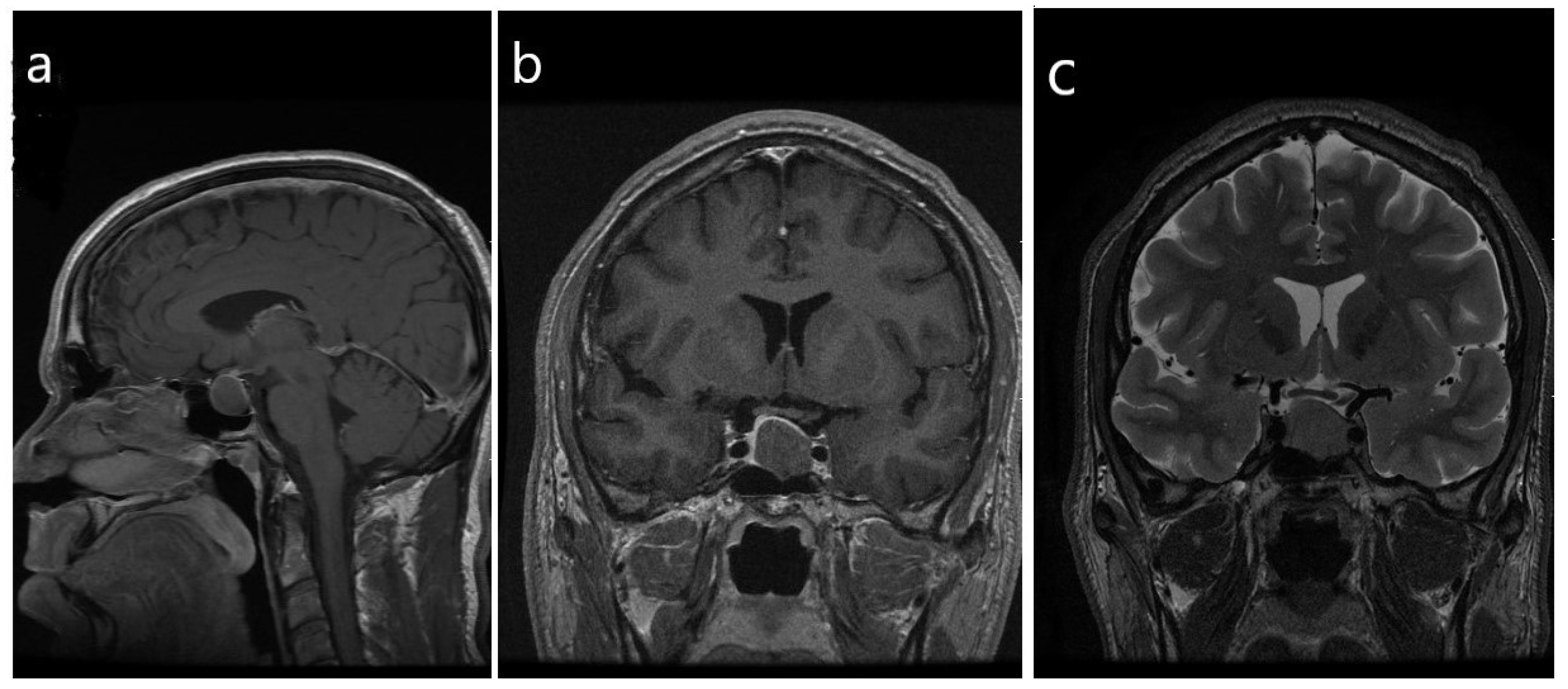
- Hypothalamic tumors known as gangliocytomas are very rare tumors that may produce excess GHRH; these benign, slow-growing, neuronal neoplasms are usually diagnosed in children and young adults. Puchner et al. reported that acromegaly was found frequently in the cases of sellar gangliocytoma; in this setting, the neuronal tumor was associated with somatotroph proliferations, either hyperplasia or, most often, sparsely-granulated somatotroph tumors [4,5].
- Ectopic GHRH secretion, causing pituitary somatotroph hyperplasia, is a rare cause of acromegaly, responsible for less than 1% of cases [3,65,76]. This phenomenon has been described in neuroendocrine tumors of pancreas or lung and pheochromocytomas; some of the patients had had familial syndromes, such as MEN1 [77,78]. Even with prolonged stimulation, there is usually hyperplasia that is thought to be reversible [79,80], however transition to the pituitary tumor has been reported in a case where there was metastasis of the lesion to the pituitary [81].
4. Conclusions
Author Contributions
Conflicts of Interest
References
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Acromegaly. N. Engl. J. Med. 2006, 355, 2558–2573. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Asa, S.L.; Kovacs, K. Growth Hormone-Releasing Hormone-Producing Tumors: Clinical, Biochemical, and Morphological Manifestations. Endocr. Rev. 1988, 9, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Bilbao, J.M.; Horvath, E.; Ryan, N.; Kovacs, K.; Singer, W.; Scheithauer, B.W.; Randall, R.V.; Laws, E.R.; Linfoot, J.A.; et al. A case for hypothalamic acromegaly: A clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J. Clin. Endocrinol. Metab. 1984, 58, 796–803. [Google Scholar] [CrossRef]
- Puchner, M.J.A.; Kudecke, D.K.; Saeger, W.; Riedel, M.; Asa, S.L. Gangliocytomas of the sellar region—A review. Exp. Clin. Endocrinol. Diabetes 1995, 103, 129–149. [Google Scholar] [CrossRef]
- Sabino, S.M.; Miranda, P.A.C.; Ribeiro-Oliveira, A. Growth hormone-secreting pituitary adenomas: From molecular basis to treatment options in acromegaly. Cancer Biol. Ther. 2010, 9, 483–492. [Google Scholar] [CrossRef]
- Ribeiro-Oliveira, A.; Barkan, A. The changing face of acromegalyg - Advances in diagnosis and treatment. Nat. Rev. Endocrinol. 2012, 8, 605–611. [Google Scholar] [CrossRef]
- Leontiou, C.A.; Gueorguiev, M.; Van Der Spuy, J.; Quinton, R.; Lolli, F.; Hassan, S.; Chahal, H.S.; Igreja, S.C.; Jordan, S.; Rowe, J.; et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. 2008, 93, 2390–2401. [Google Scholar] [CrossRef]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.-H.; et al. Gigantism and Acromegaly Due to Xq26 Microduplications and GPR101 Mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Asa, S.L. Tumors of the Pituitary Gland; American Registry of Pathology in collaboration with the Armed Forces Institute of Pathology: Washington, DC, USA, 2011. [Google Scholar]
- Asa, S.L.; Kucharczyk, W.; Ezzat, S. Pituitary Acromegaly: Not one disease Sylvia. Endocr. Relat. Cancer 2017, 24, C1–C4. [Google Scholar] [CrossRef]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Beatriz, S. Growth hormone-secreting adenomas: Pathology and cell biology. Neurosurg. Focus 2010, 29, E2. [Google Scholar] [CrossRef] [PubMed]
- Horvath, E.; Kovacs, K. Pathology of acromegaly. Neuroendocrinology 2006, 83, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Fougner, S.L.; Casar-Borota, O.; Heck, A.; Berg, J.P.; Bollerslev, J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analog treatment in a large series of patients with acromegaly. Clin. Endocrinol. 2012, 76, 96–102. [Google Scholar] [CrossRef]
- Kiseljak-Vassiliades, K.; Carlson, N.E.; Borges, M.T.; Kleinschmidt-DeMasters, B.K.; Lillehei, K.O.; Kerr, J.M.; Wierman, M.E. Growth hormone tumor histological subtypes predict response to surgical and medical therapy. Endocrine 2015, 49, 231–241. [Google Scholar] [CrossRef]
- Brzana, J.; Yedinak, C.G.; Gultekin, S.H.; Delashaw, J.B.; Fleseriu, M. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: A large single center experience. Pituitary 2013, 16, 490–498. [Google Scholar] [CrossRef]
- Kato, M.; Inoshita, N.; Sugiyama, T.; Tani, Y.; Shichiri, M.; Sano, T.; Yamada, S.; Hirata, Y. Differential expression of genes related to drug responsiveness between sparsely and densely-granulated somatotroph adenomas. Endocr. J. 2012, 59, 221–228. [Google Scholar] [CrossRef]
- Larkin, S.; Reddy, R.; Karavitaki, N.; Cudlip, S.; Wass, J.; Ansorge, O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naïve patients with somatotroph adenomas. Eur. J. Endocrinol. 2013, 168, 491–499. [Google Scholar] [CrossRef]
- Hagiwara, A.; Inoue, Y.; Wakasa, K.; Haba, T.; Tashiro, T.; Miyamoto, T. Comparison of Growth Hormone–producing and Non–Growth Hormone–producing Pituitary Adenomas: Imaging Characteristics and Pathologic Correlation. Radiology 2003, 228, 533–538. [Google Scholar] [CrossRef]
- Ezzat, S.; Caspar-Bell, G.M.; Chik, C.L.; Denis, M.-C.; Domingue, M.-È.; Imran, S.A.; Johnson, M.D.; Lochnan, H.A.; Grégoire Nyomba, B.L.; Prebtani, A.; et al. Predictive Markers for Postsurgical Medical Management of Acromegaly: A Systematic Review and Consensus Treatment Guideline. Endocr. Pract. 2019, 25, 379–393. [Google Scholar] [CrossRef]
- Ezzat, S.; Kontogeorgos, G.; Redelmeier, D.A.; Horvath, E.; Harris, A.G.; Kovacs, K. In vivo responsiveness of morphological variants of growth hormone-producing pituitary adenomas to octreotide. Eur. J. Endocrinol. 1995, 133, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Bhayana, S.; Booth, G.L.; Asa, S.L.; Kovacs, K.; Ezzat, S. The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J. Clin. Endocrinol. Metab. 2005, 90, 6290–6295. [Google Scholar] [CrossRef] [PubMed]
- Landis, A.C.; Masters, B.S.; Spada, A.; Pace, M.A.; Bourne, R.H.; Vallar, L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Vallar, L.; Spada, A.; Giannattasio, G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 1987, 330, 566–568. [Google Scholar] [CrossRef]
- Spada, A.; Arosio, M.; Bochicchio, D.; Bazzoni, N.; Vallar, L.; Bassetti, M. Patients Bearing Growth Hormone-Secreting Pituitary Tumors with or without Constitutively Active Adenylyl Cyclase. J. Clin. Endocrinol. Metab. 1990, 71, 1421–1426. [Google Scholar] [CrossRef]
- Asa, S.L.; Ezzat, S. The pathogenesis of pituitary tumours. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 97–126. [Google Scholar] [CrossRef]
- Xu, B.; Sano, T.; Yoshimoto, K.; Yamada, S. Downregulation of E-cadherin and its undercoat proteins in pituitary growth hormone cell adenomas with prominent fibrous bodies. Endocr. Pathol. 2002, 13, 341–351. [Google Scholar] [CrossRef]
- Obari, A.; Sano, T.; Ohyama, K.; Kudo, E.; Qian, Z.R.; Yoneda, A.; Rayhan, N.; Mustafizur Rahman, M.; Yamada, S. Clinicopathological features of growth hormone-producing pituitary adenomas: Difference among various types defined by cytokeratin distribution pattern including a transitional form. Endocr. Pathol. 2008, 19, 82–91. [Google Scholar] [CrossRef]
- Mete, O.; Citosun, A.; Pressman, I.; Asa, S.L. Epidemiology and biomarker profile of pituitary adenohypophysial tumors.pdf. Mod. Pathol. Pathol. 2018, 31, 900–909. [Google Scholar] [CrossRef]
- Bakhtiar, Y.; Hirano, H.; Arita, K.; Yunoue, S.; Fujio, S.; Tominaga, A.; Sakoguchi, T.; Sugiyama, K.; Kurisu, K.; Yasufuku-Takano, J.; et al. Relationship between cytokeratin staining patterns and clinico-pathological features in somatotropinomae. Eur. J. Endocrinol. 2010, 163, 531–539. [Google Scholar] [CrossRef]
- Nagata, Y.; Inoshita, N.; Fukuhara, N.; Yamaguchi-Okada, M.; Nishioka, H.; Iwata, T.; Yoshimoto, K.; Yamada, S. Growth hormone-producing pituitary adenomas in childhood and young adulthood: Clinical features and outcomes. Pituitary 2018, 21, 1–9. [Google Scholar] [CrossRef]
- Syro, L.V.; Rotondo, F.; Serna, C.A.; Ortiz, L.D.; Kovacs, K. Pathology of GH-producing pituitary adenomas and GH cell hyperplasia of the pituitary. Pituitary 2017, 20, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Heck, A.; Ringstad, G.; Fougner, S.L.; Casar-Borota, O.; Nome, T.; Ramm-Pettersen, J.; Bollerslev, J. Intensity of pituitary adenoma on T2-weighted magnetic resonance imaging predicts the response to octreotide treatment in newly diagnosed acromegaly. Clin. Endocrinol. 2012, 77, 72–78. [Google Scholar] [CrossRef]
- Heck, A.; Emblem, K.E.; Casar-Borota, O.; Bollerslev, J.; Ringstad, G. Quantitative analyses of T2-weighted MRI as a potential marker for response to somatostatin analogs in newly diagnosed acromegaly. Endocrine 2016, 52, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Potorac, I.; Petrossians, P.; Daly, A.F.; Alexopoulou, O.; Borot, S.; Sahnoun-Fathallah, M.; Castinetti, F.; Devuyst, F.; Jaffrain-Rea, M.L.; Briet, C.; et al. T2-weighted MRI signal predicts hormone and tumor responses to somatostatin analogs in acromegaly. Endocr. Relat. Cancer 2016, 23, 871–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martucci, F.; Trivellin, G.; Korbonits, M. Familial isolated pituitary adenomas: An emerging clinical entity. J. Endocrinol. Investig. 2012, 35, 1003–1014. [Google Scholar] [CrossRef]
- Dénes, J.; Kasuki, L.; Trivellin, G.; Colli, L.M.; Takiya, C.M.; Stiles, C.E.; Barry, S.; De Castro, M.; Gadelha, M.R.; Korbonits, M. Regulation of aryl hydrocarbon receptor interacting protein (AIP) protein expression by MiR-34a in sporadic somatotropinomas. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Asa, S.L.; DiGiovanni, R.; Jiang, J.; Ward, M.L.; Loesch, K.; Yamada, S.; Sano, T.; Yoshimoto, K.; Frank, S.J.; Ezzat, S. A growth hormone receptor mutation impairs growth hormone autofeedback signaling in pituitary tumors. Cancer Res. 2007, 67, 7505–7511. [Google Scholar] [CrossRef] [Green Version]
- Kiseljak-Vassiliades, K.; Xu, M.; Mills, T.S.; Smith, E.E.; Silveira, L.J.; Lillehei, K.O.; Kerr, J.M.; Kleinschmidt-DeMasters, B.K.; Wierman, M.E. Differential somatostatin receptor (SSTR) 1-5 expression and downstream effectors in histologic subtypes of growth hormone pituitary tumors. Mol. Cell. Endocrinol. 2015, 417, 73–83. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Carlsen, E.; Lugli, F.; Chiloiro, S.; Piacentini, S.; Bianchi, A.; Giampietro, A.; Mormando, M.; Clear, J.A.; Doglietto, F.; et al. Factors predicting pasireotide responsiveness in somatotroph pituitary adenomas resistant to first-generation somatostatin analogs: An immunohistochemical study. Eur. J. Endocrinol. 2015, 174, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.; Buslei, R.; Theodoropoulou, M.; Stalla, G.K.; Buchfelder, M.; Schofl, C. Molecular and functional properties of densely and sparsely-granulated GH-producing pituitary adenomas. Eur. J. Endocrinol. 2013, 169, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profil. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Mazal, P.; Czech, T.; Sedivy, R.; Aichholzer, M.; Wanschitz, J.; Klupp, N.; Budka, H. Prognostic relevance of intracytoplasmic cytokeratin pattern, hormone expression profile, and cell proliferation in pituitary adenomas of akromegalic patients. Clin. Neuropathol. 2001, 20, 163–171. [Google Scholar] [PubMed]
- Cuevas-Ramos, D.; Carmichael, J.D.; Cooper, O.; Bonert, V.S.; Gertych, A.; Mamelak, A.N.; Melmed, S. A structural and functional acromegaly classification. J. Clin. Endocrinol. Metab. 2015, 100, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Vance, M.L.; Lopes, M.B.; Xu, Z.; Chen, C.J.; Sheehan, J. Stereotactic radiosurgery for acromegaly: Outcomes by adenoma subtype. Pituitary 2015, 18, 326–334. [Google Scholar] [CrossRef]
- Lv, L.; Jiang, Y.; Yin, S.; Hu, Y.; Chen, C.; Ma, W.; Jiang, S.; Zhou, P. Mammosomatotroph and mixed somatotroph-lactotroph adenoma in acromegaly: A retrospective study with long-term follow-up. Endocrine 2019, 66, 310–318. [Google Scholar] [CrossRef]
- Asa, S.L.; Kovacs, K.; Horvath, E.; Singer, W.; Smyth, H. Hormone secretion in vitro by plurihormonal pituitary adenomas of the acidophil cell line. J. Clin. Endocrinol. Metab. 1992, 75, 68–75. [Google Scholar]
- Mete, O.; Kefeli, M.; Çalışkan, S.; Asa, S.L. GATA3 immunoreactivity expands the transcription factor profile of pituitary neuroendocrine tumors. Mod. Pathol. 2019, 32, 484–489. [Google Scholar] [CrossRef]
- Kreutzer, J.; Vance, M.L.; Lopes, M.S.; Laws, E.R. Surgical management of GH-secreting pituitary adenomas: An outcome study using modern remission criteria. J. Clin. Endocrinol. Metab. 2001, 86, 4072–4077. [Google Scholar] [CrossRef]
- Rick, J.; Jahangiri, A.; Flanigan, P.M.; Chandra, A.; Kunwar, S.; Blevins, L.; Aghi, M.K. Growth hormone and prolactin-staining tumors causing acromegaly: A retrospective review of clinical presentations and surgical outcomes. J. Neurosurg. 2018, 131, 1–7. [Google Scholar] [CrossRef]
- Sherlock, M.; Fernandez-Rodriguez, E.; Alonso, A.A.; Reulen, R.C.; Ayuk, J.; Clayton, R.N.; Holder, G.; Sheppard, M.C.; Bates, A.; Stewart, P.M. Medical therapy in patients with acromegaly: Predictors of response and comparison of efficacy of dopamine agonists and somatostatin analogs. J. Clin. Endocrinol. Metab. 2009, 94, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandret, L.; Maison, P.; Chanson, P. Place of cabergoline in acromegaly: A meta-analysis. J. Clin. Endocrinol. Metab. 2011, 96, 1327–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, E.; Kovacs, K.; Singer, W.; Smyth, H.S.; Killinger, D.W.; Erzin, C.; Weiss, M.H. Acidophil stem cell adenoma of the human pituitary: Clinicopathologic analysis of 15 cases. Cancer 1981, 47, 761–771. [Google Scholar] [CrossRef]
- Gomez-Hernandez, K.; Ezzat, S.; Asa, S.L.; Mete, Ö. Clinical implications of accurate subtyping of pituitary adenomas: Perspectives from the treating physician. Turk Patoloji Derg. 2015, 31, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Gomez-Hernandez, K.; Kucharczyk, W.; Ridout, R.; Zadeh, G.; Gentili, F.; Ezzat, S.; Asa, S.L. Silent subtype 3 pituitary adenomas are not always silent and represent poorly differentiated monomorphous plurihormonal Pit-1 lineage adenomas. Mod. Pathol. 2016, 29, 131–142. [Google Scholar] [CrossRef]
- Alshaikh, O.M.; Asa, S.L.; Mete, O.; Ezzat, S. An Institutional Experience of Tumor Progression to Pituitary Carcinoma in a 15-Year Cohort of 1055 Consecutive Pituitary Neuroendocrine Tumors. Endocr. Pathol. 2019, 30, 118–127. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.; Kloppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs, 4th ed.; IARC Press: Lyon, France, 2017. [Google Scholar]
- Asa, S.L.; Ezzat, S. Aggressive Pituitary Tumors or Localized Pituitary Carcinomas: Defining Pituitary Tumors. Expert Rev. Endocrinol. Metab. 2016, 11, 149–162. [Google Scholar] [CrossRef]
- Thapar, K.; Kovacs, K.; Scheithauer, B.W.; Stefaneanu, L.; Horvath, E.; Pernicone, P.J.; Murray, D.; Laws, E.R. Proliferative activity and invasiveness among pituitary adenomas and carcinomas: An analysis using the MIB-1 antibody. Neurosurgery 1996, 38, 99–107. [Google Scholar] [CrossRef]
- Dudziak, K.; Honegger, J.; Bornemann, A.; Horger, M.; Müssig, K. Pituitary carcinoma with malignant growth from first presentation and fulminant clinical course—Case report and review of the literature. J. Clin. Endocrinol. Metab. 2011, 96, 2665–2669. [Google Scholar] [CrossRef] [Green Version]
- Sreenan, S.; Sengupta, E.; Tormey, W.; Landau, R. Metastatic pituitary carcinoma in a patient with acromegaly: A case report. J. Med. Case Rep. 2012, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Greenman, Y.; Pei, L.; Melmed, S.; Said, J.W.; Woolf, P.; Coniglio, J. Remission of acromegaly caused by pituitary carcinoma after surgical excision of growth hormone-secreting metastasis detected by 111-indium pentetreotide scan. J. Clin. Endocrinol. Metab. 1996, 81, 1628–1633. [Google Scholar] [PubMed] [Green Version]
- Le Roux, W.C.; Mulla, A.; Meeran, K. Pituitary Carcinoma as a Cause of Acromegaly. N. Engl. J. Med. 2001, 345, 1645–1646. [Google Scholar] [CrossRef] [PubMed]
- Garby, L.; Caron, P.; Claustrat, F.; Chanson, P.; Tabarin, A.; Rohmer, V.; Arnault, G.; Bonnet, F.; Chabre, O.; Christin-Maitre, S.; et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of Growth Hormone-Releasing Hormone (GHRH): A French nationwide series of 21 cases. J. Clin. Endocrinol. Metab. 2012, 97, 2093–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trouillas, J.; Labat-Moleur, F.; Sturm, N.; Kujas, M.; Heymann, M.F.; Figarella-Branger, D.; Patey, M.; Mazucca, M.; Decullier, E.; Vergès, B.; et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am. J. Surg. Pathol. 2008, 32, 534–543. [Google Scholar] [CrossRef]
- Schernthaner-Reiter, M.H.; Trivellin, G.; Stratakis, C.A. MEN1, MEN4, and Carney Complex: Pathology and Molecular Genetics. Neuroendocrinology 2016, 103, 18–31. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Caswell, R.; Bunce, B.; Jose, S.; Yuan, B.; Hernández-Ramírez, L.C.; Kapur, S.; Caimari, F.; Evanson, J.; Ferraù, F.; et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol. Commun. 2016, 4, 56. [Google Scholar] [CrossRef]
- Salenave, S.; Boyce, A.M.; Collins, M.T.; Chanson, P. Acromegaly and mccune-albright syndrome. J. Clin. Endocrinol. Metab. 2014, 99, 1955–1969. [Google Scholar] [CrossRef] [Green Version]
- Krug, S.; Boch, M.; Rexin, P.; Pfestroff, A.; Gress, T.; Michl, P.; Rinke, A. Acromegaly in a patient with a pulmonary neuroendocrine tumor: Case report and review of current literature. BMC Res. Notes 2016, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Beuschelin, F.; Strasburger, J.C.; Siegerstetter, V.; Mordapour, D.; Lichter, P.; Bidlingmaier, M.; Blum, E.H.; Reincke, M. Acromegaly Caused by Secretion of Growth Hormone by A Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2000, 342, 1871–1876. [Google Scholar] [CrossRef]
- Melmed, S.; Ezrin, C.; Kovacs, K.; Goodman, S.R.; Forhman, A.L. Acromegaly Due To Secretion of Growth Hormone By An Ectopic Pancreatic Islet-Cell Tumor. N. Engl. J. Med. 1988, 312, 9–16. [Google Scholar] [CrossRef]
- Ezzat, S.; Yamashita, S.; Melmed, S. Recurrent acromegaly resulting from ectopic growth hormone gene expression by a metastatic pancreatic tumor. Cancer 1993, 71, 66–70. [Google Scholar] [CrossRef]
- Hori, E.; Akai, T.; Kurimoto, M.; Hirashima, Y.; Endo, S. Growth hormone-secreting pituitary adenoma confined to the sphenoid sinus associated with a normal-sized empty sella. J. Clin. Neurosci. 2002, 9, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.; Hernández-Ramirez, L.C.; Espinosa-De-Los-Monteros, A.L.; Franco, J.M.; Guinto, G.; Mercado, M. Ectopic acromegaly due to a GH-secreting pituitary adenoma in the sphenoid sinus: A case report and review of the literature. BMC Res. Notes 2013, 6, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losa, M.; Schopohl, J.; von Werder, K. Ectopic secretion of growth hormone-releasing hormone (GHRH). J. Endocrinol. Investig. 1993, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Sala, E.; Ferrante, E.; Verrua, E.; Malchiodi, E.; Mantovani, G.; Filopanti, M.; Ferrero, S.; Pietrabissa, A.; Vanoli, A.; La Rosa, S.; et al. Growth hormone-releasing hormone-producing pancreatic neuroendocrine tumor in a multiple endocrine neoplasia type 1 family with an uncommon phenotype. Eur. J. Gastroenterol. Hepatol. 2013, 25, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Othman, N.H.; Asa, S.L.; Ezzat, S.; Kovacs, K.; Horvath, E.; Poulin, E.; Smyth, H.S. Growth hormone-releasing hormone (GHRH) and GHRH receptor (GHRH-R) isoform expression in ectopic acromegaly. Clin. Endocrinol. 2001, 55, 135–140. [Google Scholar] [CrossRef]
- Ezzat, S.; Asa, S.L.; Stefaneanu, L.; Whittom, R.; Smyth, H.S.; Horvath, E.; Kovacs, K.; Frohman, L.A. Somatotroph Hyperplasia without Pituitary Adenoma Associate&with a Long Standing Growth Hormone- Releasing Hormone-Producing Bronchial Carcinoid. J. Clin. Endocrinol. Metab. 1994, 78, 555–560. [Google Scholar]
- Ramsay, J.A.; Kovacs, K.; Asa, S.L.; Pike, M.J.; Thorner, M.O. Reversible sellar enlargement due to growth hormone-releasing hormone production by pancreatic endocrine tumors in an acromegalic patient with multiple endocrine neoplasia type I syndrome. Cancer 1988, 62, 445–450. [Google Scholar] [CrossRef]
- Nasr, C.; Mason, A.; Mayberg, M.; Staugaitis, S.M.; Asa, S.L. Acromegaly and somatotroph hyperplasia with adenomatous transformation due to pituitary metastasis of a growth hormone-releasing hormone-secreting pulmonary endocrine carcinoma. J. Clin. Endocrinol. Metab. 2006, 91, 4776–4780. [Google Scholar] [CrossRef]
- Borson-Chazot, F.; Garby, L.; Raverot, G.; Claustrat, F.; Raverot, V.; Sassolas, G. Acromegaly induced by ectopic secretion of GHRH: A review 30 years after GHRH discovery. Ann. Endocrinol. 2012, 73, 497–502. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akirov, A.; Asa, S.L.; Amer, L.; Shimon, I.; Ezzat, S. The Clinicopathological Spectrum of Acromegaly. J. Clin. Med. 2019, 8, 1962. https://doi.org/10.3390/jcm8111962
Akirov A, Asa SL, Amer L, Shimon I, Ezzat S. The Clinicopathological Spectrum of Acromegaly. Journal of Clinical Medicine. 2019; 8(11):1962. https://doi.org/10.3390/jcm8111962
Chicago/Turabian StyleAkirov, Amit, Sylvia L. Asa, Lama Amer, Ilan Shimon, and Shereen Ezzat. 2019. "The Clinicopathological Spectrum of Acromegaly" Journal of Clinical Medicine 8, no. 11: 1962. https://doi.org/10.3390/jcm8111962
APA StyleAkirov, A., Asa, S. L., Amer, L., Shimon, I., & Ezzat, S. (2019). The Clinicopathological Spectrum of Acromegaly. Journal of Clinical Medicine, 8(11), 1962. https://doi.org/10.3390/jcm8111962