Abstract
Gout is an inflammatory arthritis influenced by environmental risk factors and genetic variants. The common dysfunctional p.Q141K allele of the ABCG2 gene affects gout development. We sought after the possible association between the p.Q141K variant and gout risk factors, biochemical, and clinical determinants in hyperuricemic, gouty, and acute gouty arthritis cohorts. Further, we studied the correlation of p.Q141K allele and levels of pro-/anti-inflammatory cytokines. Coding regions of the ABCG2 gene were analyzed in 70 primary hyperuricemic, 182 gout patients, and 132 normouricemic individuals. Their genotypes were compared with demographic and clinical parameters. Plasma levels of 27 cytokines were determined using a human multiplex cytokine assay. The p.Q141K variant was observed in younger hyperuricemic/gout individuals (p = 0.0003), which was associated with earlier disease onset (p = 0.004), trend toward lower BMI (p = 0.056), and C-reactive protein (CRP, p = 0.007) but a higher glomerular filtration rate (GFR, p = 0.035). Levels of 19 cytokines were higher, mainly in patients with acute gouty arthritis (p < 0.001), irrespective of the presence of the p.Q141K variant. The p.Q141K variant influences the age of onset of primary hyperuricemia or gout and other disease-linked risk factors and symptoms. There was no association with cytokine levels in the circulation.
1. Introduction
Gout is the most common type of inflammatory arthritis in adults that develops as a consequence of elevated urate levels. Gout has four phases: asymptomatic hyperuricemia, acute gouty arthritis, intercritical gout, and chronic tophaceous gout [1]. The attacks are caused by an inflammatory response to monosodium urate (MSU) crystals that deposit in joints, tendons, and surrounding tissues. Before the development of gout, patients show asymptomatic hyperuricemia (elevated levels of serum uric acid >420 µmol/L for men and >360 µmol/L for women). Not all individuals with hyperuricemia develop symptomatic gout, but the risk increases in proportion to the elevation of urate in circulation. Heritability of serum urate levels and gout in Europeans has been estimated to be approximately 30% [2].
The mechanism of gouty inflammation is coupled with the formation and activation of the NOD-like receptor P3 (NLRP3) inflammasome with subsequent production of pro-inflammatory cytokines. MSU crystals by themselves are not responsible for the induction of expression and assembly of inflammasome components. The first signal involves priming monocyte-derived macrophages, which then start binding ligands (S100A8 and S100A9 proteins, long-chain free fatty acids) to Toll-like receptors (TLR) [3]. The second signal, triggered by MSU crystals, leads to activation of multiprotein intracellular NLRP3 inflammasomes, which contain pro-caspase 1 [4]. Next, active caspase 1 cleaves precursors of interleukins 1 beta and 18 (pro-IL-1β and pro-IL-18) to generate the active forms (IL-1β and IL-18) [5,6]. Secretion of IL-1β leads to recruitment of neutrophils to the site of inflammation, production of additional pro-inflammatory cytokines, and bone/cartilage degradation [3,7]. IL-1β and IL-18 induced other pro-inflammatory cytokines IL-6, IL-8, IL-17, and tumor necrosis factor alpha (TNFα), which synergistically potentiate gouty inflammation [8,9,10].
Risk of gout development is conditioned not only by hyperuricemia but also by gender, weight, age, environmental and genetic factors, and their interactions. The main cause of hyperuricemia is a defect in renal excretion of urate. Today 10 genes for the main urate transporters are known, including ATP-binding cassette subfamily G member 2 (ABCG2), which has the greatest influence on urate excretion. The ABCG2 protein is a homodimeric membrane transporter with functions in a variety of tissues, including xenobiotic transport. Many population-specific variants of the ABCG2 gene have been found e.g., the common non-synonymous variants p.Q126X (rs72552713), p.V12M (rs2231137), and p.Q141K (rs2231142) [11,12]. The p.Q126X variant is specific to East Asian populations and has a well-known genetic impact on gout induction. It decreases ABCG2 urate transport by 96%. A second common variant in Caucasians is p.V12M, which showed a protective role in gout development and has no influence on urate transport in human embryonic kidney derived cells [13,14]. The missense p.Q141K variant (minor frequency allele (MAF), present in the Central European Caucasian population at around 9.4%) is localized in the nucleotide-binding domain and leads to decreased binding of adenosine triphosphate (ATP) and reduces transport function by almost 50% [14,15,16]. Carriers of p.Q141K tend to have a higher chance of developing an earlier gout onset and have an increased risk of a poor response to allopurinol [12,17,18,19,20]. Moreover, p.Q141K may also influence progression from asymptomatic hyperuricemia to gout by promoting the immune response to MSU crystals. This suggests a hypothetic molecular pathway that connects non-functional variants of urate transporters and the innate immune response [1]. In previous studies, pro-inflammatory cytokines affected ABCG2 gene expression. Its expression could be upregulated or downregulated by IL-1β and TNFα treatment, either alone or in combination with other biologically active molecules such as estrogen [21,22,23].
In our previous studies using an ABCG2 analysis of Czech patients with hyperuricemic/gout, we found that both the rare and common p.Q141K variants are independently associated with hyperuricemia and gout [12,13]. Moreover, we found a significantly higher frequency of the minor allele p.Q141K variant in pediatric-onset patients when compared to adult-onset patients [24]. Our latest published study demonstrated the effects of rare variants on the expression, cellular localization, and function of ABCG2 [25].
The aim of this study was to analyze (1) the association of the p.Q141K variant with known risk factors of gout (e.g., age, BMI, etc.), (2) the possible link between the risk of the p.Q141K allele and levels of biochemical parameters connected with gout (CRP, estimated glomerular filtration rate calculated using the Modification of Diet in Renal Disease (eGFR-MDRD)), and finally (3) the relationship between the p.Q141K allele and pro-/anti-inflammatory cytokines using human multiplex cytokine assay in patients with primary hyperuricemia, gout, acute gouty arthritis, and normouricemic individuals. Relationships between the p.Q141K variant and cytokine plasma levels were addressed for the first time.
2. Experimental Section
2.1. Cohort and Subcohorts
The study cohort comprised 182 patients with primary gout (166 males, 16 females; median age = 54 years), including 17 patients (23 samples) having a gout attack, and 70 patients with asymptomatic hyperuricemia (50 males, 20 females; median age = 38 years) from the biobank of the Institute of Rheumatology, Prague, the Czech Republic. Gout patients met the 1977 American Rheumatism Association preliminary classification criteria [26]. Primary hyperuricemic patients were classified as serum uric acid (SUA) >420 µmol/L for men and SUA > 360 µmol/L for women on two measurements. Two repetitive measurements of purine metabolites (xanthine, hypoxanthine, and oxypurinol), serum uric acid (SUA), and biochemical parameters were performed to exclude the impact of purine metabolic disorders in all patients. The first xanthine, hypoxanthine, and oxypurinol metabolites, SUA, and biochemical determination were performed during allopurinol/febuxostat treatment. The second measurement was done before the start of treatment or 72 h after a temporary interruption of SUA lowering therapy. Patients were compared to 132 normouricemic individuals from the general population, with no history of primary hyperuricemia, gout, or autoimmune disease (54 males, 78 females; median age = 41 years). All patients and normouricemic subjects were examined for the functional p.Q141K variant of the ABCG2 gene, as published previously [13]. All demographic (age of disease onset and age of examination/sampling, sex, body mass index (BMI)), biochemical (SUA with/off treatment, fractional excretion of uric acid (FE-UA), estimated glomerular filtration rate calculated using the Modification of Diet in Renal Disease formula (eGFR-MDRD), serum creatinine, and maximal C-reactive protein (CRP)), genetic (familial occurrence, presence of the wild-type p.Q141K variant (GG), heterozygotic (GT), and homozygotic (TT) form and its MAF) and presence and type of medical treatment characteristics of patients and normouricemic individuals are stated in Table 1. This study was approved by the local ethics committee, and all patients and normouricemic individuals signed informed consent.

Table 1.
Main demographic, biochemical, and genetic characteristics of the normouricemic (n = 132), hyperuricemic (n = 70), and gout-suffering subjects (n = 182).
2.2. Cytokine Detection
In total, 27 cytokines (IL-1β, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12, IL-13, IL-15, IL-17, IP-10, MCP-1, FGF-basic, eotaxin, G-CSF, GM-CSF, IFN-γ, MIP-1α, MIP-1β, PDGF, RANTES, TNF-α, and VEGF) were determined in 149 selected plasma samples from 132 (out of 182) gout patients, in 23 samples from 17 gout patients during an inflammatory attack, in 44 (out of 70) hyperuricemic patients, and in 132 normouricemic subjects. Samples were tested using commercial Bio-Plex Pro TM Human Cytokine 27-plex Assay kits (Bio-Rad, California, USA) and a Bio-Plex 200 system (Bio-Rad, California, USA). Plasma samples were prepared using a standard protocol and aliquoted to storage at −80 °C until analyzed.
2.3. Statistical Analysis
Data were summarized as means with standard deviations (SD) and/or medians with interquartile ranges (IQR) where appropriate. Continuous subject characteristics between groups were compared using the Kruskal–Wallis ANOVA, and categorical characteristics were compared using the Fisher exact test. Associations between characteristics of the diagnostic group (i.e., with the normouricemic, hyperuricemic, or gouty status) and the p.Q141K allele dose were modeled using linear regression with interactions; creatinine and CRP values were log-transformed for a better fit. Associations between cytokine measurements and diagnostic groups and the p.Q141K allele dose were modeled using linear mixed models with group and allele dose as fixed effects and subject ID as the random factor since some individuals were measured multiple times. All cytokine measurements, except for eotaxin, were log-transformed for a better fit. The Benjamini–Hochberg multiple comparisons correction was used where appropriate. All analyses were performed using statistical language and environment R, version 3.4.4.
3. Results
3.1. Analysis of the p.Q141K Variant in Normouricemic and Patient Cohort
First, we analyzed the presence of the p.Q141K allele in the ABCG2 gene in our study cohort. From 132 normouricemic subjects with an MAF = 7.1%, 15 were heterozygote, and one was homozygote for p.Q141K variant. In the hyperuricemic cohort with an MAF = 21.3%, 19 were heterozygous, and five were homozygous for the p.Q141K allele. Finally, in gout patients with an MAF = 25%, 68 patients were GT heterozygotes and 11 were TT homozygotes. Gout and hyperuricemic patients had significantly more heterozygous and homozygous variants and hence, a significantly higher p.Q141K frequency than normouricemic subjects (p < 0.0001) (Table 1).
3.2. Analysis of the p.Q141K Variant and Its Relationship with Risk Factors for Gout
Age, BMI, CRP, and GFR are known risk factors for gout. For each of these, we found expected differences between the diagnostic groups but their association with a diagnostic status varied depending on the p.Q141K allele dose.
Risk of gout and hyperuricemia increases proportionally with age. In our cohort, patients with hyperuricemia or gout were older than normouricemic individuals (p < 0.0001, Figure 1A) during the time of the study. However, hyperuricemic and gout patients homozygous for p.Q141K were significantly younger than wild-types and heterozygotes (p = 0.0003). Furthermore, hyperuricemia and gout manifested earlier in p.Q141K homozygotes (p = 0.004, Figure 1B).
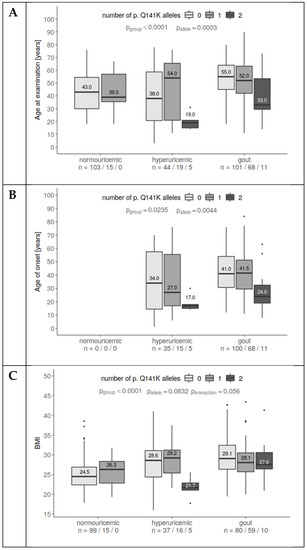
Figure 1.
The association of p.Q141K alleles with age at examination, age of onset, and BMI. (A) Gout patients were older than hyperuricemic and normouricemic subjects. Homozygotes for p.Q141K were younger than wild type/heterozygotes. Interactions suggest different associations for normouricemic subjects. (B) There are no substantial differences in the age of onset between hyperuricemia and gout individuals. p.Q141K homozygotes tend to have much earlier disease onset than others. (C) Gout and hyperuricemic patients had higher BMIs than controls. Homozygous patients were slightly leaner than others. A statistically significant interaction suggests different associations for p.Q141K among groups. This may have been influenced by the pediatric hyperuricemia subset.
Elevated BMI increases the probability of metabolic syndrome as well as the probability of hyperuricemia and gout. This is in line with our data: Patients with hyperuricemia or gout had a higher BMI than normouricemic patients (p < 0.0001). In contrast, homozygotes for the p.Q141K variant tended to have lower BMIs (p = 0.056, Figure 1C). The homozygotes with lower BMIs were mostly very similar to those with young-age hyperuricemia onset.
Similarly, to age and BMI, CRP levels were higher in hyperuricemic and gouty patients than in normouricemic individuals (p < 0.0001). But significantly lower levels of CRP were measured in p.Q141K homozygous hyperuricemic and gout patients (p = 0.007, Figure 2A).
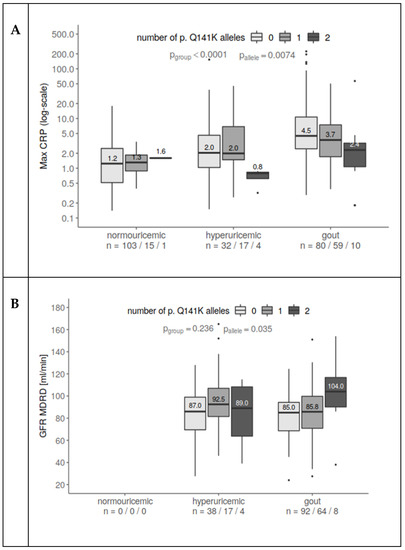
Figure 2.
The connection between the presence of p.Q141K, maximum CRP, and eGFR-MDRD. (A) CRP increased with hyperuricemia and even more for gout patients compared to normouricemic subjects. At the same time, p.Q141K homozygotes had lower CRP than heterozygotes and wild type. The relationship prevailed after including age as a covariate (CRP slightly rises with subject age). (B) GFR increases with an increasing number of p.Q141K alleles, in a similar way for both hyperuricemic and gout patients. One extreme observation (426 mL/min) was excluded since it heavily influenced the fit. All values were log-transformed for a better fit.
Although GFR should be decreased in patients with hyperuricemia and gout because of worsened renal function, heterozygous and mainly homozygous carriers of the p.Q141K variant had significantly higher GFRs (p = 0.035, Figure 2B, data for normouricemic subjects were not available).
3.3. Analysis of the p.Q141K Variant in Association with Cytokine Plasma Levels
To determine the possible differences between normouricemic, hyperuricemic, gout, and acute gout patients on immunological levels, we determined the cytokine plasma profiles of our cohort. Then, we analyzed the possible influence of the p.Q141K variant on cytokine production between patient groups.
We found that levels of IL-1β, IL-1ra, IL-4, IL-6, IL-7, IL-8, IL-9, IL-13, IL-17, fibroblast growth factor-basic (FGF-basic), interferon gamma (IFNγ), and TNFα were significantly increased in patients with acute gouty arthritis compared to other groups (multiple correction p-values ranging from p < 0.05 to p < 0.001, see Figure 3). On the other hand, eotaxin, monocyte chemoattractant protein-1 (MCP-1), and regulated on activation, normal T cell expressed and secreted (RANTES) were decreased in all patient groups compared to normouricemic individuals (multiple correction p-values ranging p < 0.001, p = 0.014, and p < 0.001, respectively). IL-5, IL-10, and IL-12 levels gradually decreased from control, subsequently hypouricemia, and gout to gout attack group. Plasma levels of IL-2, IL-15, granulocyte-colony stimulating factor (G-CSF), granulocyte-macrophage colony stimulating factor (GM-CSF), interferon gamma-induced protein-10 (IP-10), macrophage inflammatory protein-1 beta (MIP-1β), platelet derived growth factor (PDGF), and vascular endothelial growth factor (VEGF) were comparable among all groups (Figure 3, the p-values shown are after the Benjamini–Hochberg correction). We did not find any impact of p.Q141K on the plasma cytokine levels of patients or normouricemic subjects.
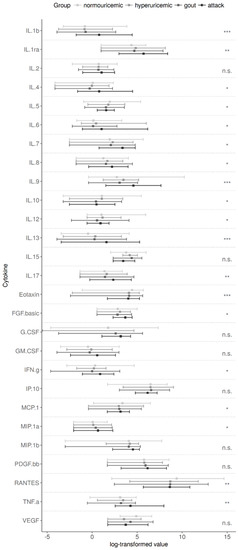
Figure 3.
Individual cytokine measurements in the compared groups. Post-hoc pairwise comparisons revealed the main source of differences in the gout attack group were: interleukin (IL)-1b, IL-1ra, IL-4, IL-6, IL-7, IL-8, IL-9, IL-13, IL-17, fibroblast growth factor (FGF)-basic, interferon gamma (IFNγ), and tumor necrosis factor alpha (TNFα). IL-5, IL-10, and IL-12 levels gradually decreased from control to gout attack group. Eotaxin, monocyte chemoattractant protein 1 (MCP-1), and regulated on activation, normal T cell expressed and secreted (RANTES) showed significantly higher values for controls. There were no associations between the p.Q141K allele and cytokine levels. p-values via an ANOVA model using log-transformed measurements (except eotaxin) are corrected for multiple comparisons (Benjamini–Hochberg method). n.s. p > 0.05, * p ≤ 0.05, ** p < 0.01, *** p < 0.001.
4. Discussion
The ABCG2 protein is a membrane-associated transporter with a wide range of functions including excretion of urate. The ABCG2 gene is expressed in the intestine, liver, kidney, and several other tissues that have a barrier function. The renal excretion of urate accounts for approximately two-thirds of total urate excretion while intestinal excretion accounts for the rest [27]. Dysfunction in this protein is particularly caused by the p.Q141K variant, which occurs at a high frequency in the Caucasian population. The variant leads to hyperuricemia and gout. Homozygotes for p.Q141K have 53% reduced excretion of urate compared to wild types [28]. Hypothetically, this allele also promotes progression from hyperuricemia to gout by stimulating the immune response to MSU crystals [1]. Therefore, we looked for a possible relationship between the number of p.Q141K alleles (i.e., wild type, heterozygous, and homozygous) and risk factors (age, BMI) or related determinants (CRP, GFR), and plasma levels of pro/anti-inflammatory cytokines.
Risk of gout increases with age, which is in line with our results where patients with hyperuricemia or gout were older than normouricemic individuals [13,29]. However, hyperuricemic and gout patients homozygous for p.Q141K were significantly younger than wild-types and heterozygotes. They also had an earlier disease onset. These findings are supported by the fact that non-functional ABCG2 transporters have a greater impact on disease progression (PAR0% = 29.2%) than risk factors such as age (PAR% = 5.74%), obesity (PAR% = 18.7%), and alcohol consumption (PAR% = 15.4%) [29].
Another risk factor for hyperuricemia and gout as well as for metabolic syndrome is increased BMI [30]. This corresponds to our data showing that patients with hyperuricemia or gout had higher BMIs than normouricemic individuals. In contrast with previously mentioned data, homozygotes for the p.Q141K variant tended to have lower BMIs. These patients were very similar to those with young-age onset. This finding suggests that the beginning of the disease was mainly influenced by the presence of p.Q141K rather than risk factors such as BMI.
In addition to the fact that high CRP levels indicate infection- or non-infection-based inflammation, serum CRP levels positively correlate with serum urate levels and can be used as a gout determinant [31]. Moreover, CRP levels increase with age and the amount of adipose tissue [32,33]. In this study, we found higher CRP levels in hyperuricemic and gouty patients than in normouricemic controls. However, significantly lower levels of CRP were found in p.Q141K homozygotes. These results show a similar trend as BMI, which could be explained by the younger age of the homozygous p.Q141K allele carriers.
GFR defines kidneys function and deteriorates in patients with hyperuricemia and gout due to worsened renal function [34]. In our study, heterozygous and mainly homozygous carriers of the p.Q141K variant had significantly higher eGFR-MDRD than patients with the wild-type variant. This finding could be explained by the gradually decreasing age of disease onset between wild-type, heterozygous, and homozygous carriers of the p.Q141K variant, although, mainly in hyperuricemic patients. Although eGFR-MDRD was not measured in normouricemic controls, all patients reached eGFR-MDRD reference levels; thus, we did not observe deteriorated renal function in our patient cohort. Another explanation for the high eGRF-MDRD could be the low mean age (53.6 years) of our study cohort compared to the mean age (61.9 years) of the gout patients described in the literature [35]. Our patients were younger, and they did not have impaired kidney function. The higher eGFR-MDRD in homozygotes might not be representative because of the low number of patients (five with hyperuricemia and seven with gout). Further experiments on a larger cohort are necessary to confirm or disprove this particular finding.
Another aim of our study was to examine the differences in cytokine levels between normouricemic, hyperuricemic, gouty, and acute gouty arthritis patients in the presence of the p.Q141K variant of the ABCG2 gene. Expression of the ABCG2 gene is impacted by pro-inflammatory cytokines and signaling cascades. One of the major transcription factors of pro-inflammatory pathways, nuclear factor kappa B (NF-κB), is able to bind to the promoter of the ABCG2 gene and upregulate it, with or without the influence of an estrogen receptor [23,36]. On the other hand, the ABCG2 gene alone is capable of inhibiting NF-κB activation, which leads to the downregulation of IL-8 [37]. Pro-inflammatory cytokines IL-1β and TNFα can have an impact on ABCG2 expression at the mRNA as well as the protein level, depending on the kind of cell line. Further, the ABCG2 protein is seen to be downregulated in colon biopsies from patients with active inflammatory bowel disease [38,39,40]. The presence of the p.Q141K variant in the ABCG2 protein leads to misfolding and decreased cell surface expression [17]. These data led us to study the impact of p.Q141K on cytokine levels. Unfortunately, we did not find any influence of this variant on cytokine protein expression in the plasma of patients from different groups or in normouricemic individuals. The likely reason our hypothesis failed was the relatively small number of patients included in the study. Further investigations using larger cohorts are needed.
Nonetheless, we found higher plasma levels of IL-1β, IL-1ra, IL-4, IL-6, IL-8, IL-9, IL-13, IL-17, FGF-basic, IFNγ, and TNFα in acute gouty arthritis patients. All these cytokines are involved in interconnected pro-/anti-inflammatory cascades, which suggest an active immune response in patients with acute gouty arthritis. On the other hand, IL-5, IL-10, and eotaxin were lower in patients with acute gout inflammation, which can be explained by their role in inhibiting inflammation (IL-10) or eosinophilic inflammation (IL-5, eotaxin) [41]. Our results regarding IL-10 levels are supported by similar findings of circulating IL-10 in a Netherlands gouty arthritis study cohort and in New Zealand patients with intercritical gout study [42]. Estevez-Garcia and colleagues measured serum levels of IL-5 among groups of normouricemic controls, hyperuricemic, and gouty patients [43] and found, in contrast to our results, IL-5 levels were the same among groups, which may be explained by IL-5 determination in serum vs. plasma. Plasma is a more sensitive matrix for detecting levels of low-abundance cytokines [44]. Eotaxin was described to be increased in obese patients with metabolic syndrome (mean BMI 38.9 ± 6.3) compared to lean subjects (mean BMI 22.4 ± 2.5) and decreased after their weight-loss [45]. This suggests that eotaxin should have been higher in our patients than in normouricemic individuals; however, our patients were not extremely obese. MCP-1 and RANTES were also lower in acute gouty arthritis patients compare to normouricemic controls in our study. However, increased serum levels of MCP-1 have been observed in hyperuricemic, gouty, and acute gouty arthritis patients, and MCP-1 has been positively correlated with elevated serum levels of uric acid [46]. The contradictory results could be explained by measurement of cytokines in plasma samples in our study. To date no association of RANTES plasma/serum levels with gout has been published; however, it is known that urate stimulates mRNA and protein expression of RANTES and MCP-1 in mice tubular epithelial cells [47]. On the other hand, synovial fluid levels of RANTES were found to be significantly lower in patients with acute gout than in patients with acute rheumatoid arthritis [48].
One of the limitations of our study was the relatively small number of p.Q141K homozygotes with hyperuricemia and gout and also a small cohort size of those with acute gouty arthritis. A larger group for p.Q141K homozygotes and acute gout arthritic patients is necessary to confirm the lower age of onset, BMI, CRP, and higher eGFR-MDRD, and cytokine levels between groups. Our results reflect the fact that our study cohort was purposefully compiled from patients with primary hyperuricemia and gout who are younger (mean age = 53.6 years) than the common gouty population (mean age = 61.9 years) and have a positive family correlation [35]. This characteristic can be considered to be one of the strengths of the study because the cohort was well-defined and excluded those with secondary hyperuricemia and gout. Our study also used explicit definitions of medication and comorbidities for the study cohorts. Finally, according to available data, and for the first time, our study examined a possible link between the p.Q141K variant and plasma cytokine levels.
5. Conclusions
In conclusion, we found that the p.Q141K variant of the ABCG2 gene impacts the age of hyperuricemia and gout onset, levels of BMI and CRP, and renal function. This variant has no effect on plasma cytokine levels of hyperuricemic and gouty patients. However, we did find higher cytokine levels, but mainly in patients with acute gouty arthritis.
Author Contributions
Conceptualization, B.S. and V.H.; methodology, B.S. and V.H.; validation, B.S. and M.P.; formal analysis and investigation, V.H., J.B., and K.P. (Kateřina Pavelcová); data curation, M.P.; writing—original draft preparation, V.H. and J.B.; writing—review and editing, B.S., L.Š., and K.P. (Karel Pavelka); visualization, M.P.; supervision, B.S. and L.Š.; project administration, B.S.; funding acquisition, B.S.
Funding
This research was funded by the Czech Ministry of Health: AZV 15-26693 A and projects for conceptual development of research organization 00023728 (Institute of Rheumatology), RVO VFN64165, SVV 260367, and SVV 260373.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Major, T.J.; Dalbeth, N.; Stahl, E.A.; Merriman, T.R. An update on the genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2018, 14, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Cadzow, M.; Merriman, T.R.; Dalbeth, N. Performance of gout definitions for genetic epidemiological studies: Analysis of UK Biobank. Arthritis Res. Ther. 2017, 19, 181. [Google Scholar] [CrossRef] [PubMed]
- So, A.K.; Martinon, F. Inflammation in gout: Mechanisms and therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 639–647. [Google Scholar] [CrossRef] [PubMed]
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef]
- Deuteraiou, K.; Kitas, G.; Garyfallos, A.; Dimitroulas, T. Novel insights into the role of inflammasomes in autoimmune and metabolic rheumatic diseases. Rheumatol. Int. 2018, 38, 1345–1354. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Schlesinger, N.; Thiele, R.G. The pathogenesis of bone erosions in gouty arthritis. Ann. Rheum. Dis. 2010, 69, 1907–1912. [Google Scholar] [CrossRef]
- Cavalcanti, N.G.; Marques, C.D.L.; Lins e Lins, T.U.; Pereira, M.C.; Rêgo, M.J.B.D.M.; Duarte, A.L.B.P.; Pitta, I.D.R.; Pitta, M.G.D.R. Cytokine profile in gout: Inflammation driven by IL-6 and IL-18? Immunol. Investig. 2016, 45, 383–395. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Q.; Yin, Y.; McNutt, M.A.; Zhang, T.; Cao, Y. Serum levels of IL-17 are elevated in patients with acute gouty arthritis. Biochem. Biophys. Res. Commun. 2018, 497, 897–902. [Google Scholar] [CrossRef]
- Mills, K.H.G.; Dungan, L.S.; Jones, S.A.; Harris, J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J. Leukoc. Biol. 2013, 93, 489–497. [Google Scholar] [CrossRef]
- Matsuo, H.; Ichida, K.; Takada, T.; Nakayama, A.; Nakashima, H.; Nakamura, T.; Kawamura, Y.; Takada, Y.; Yamamoto, K.; Inoue, H.; et al. Common dysfunctional variants in ABCG2 are a major cause of early-onset gout. Sci. Rep. 2013, 3, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Miyata, H.; Závada, J.; Tomčík, M.; Pavelka, K.; Storkanova, G.; Toyoda, Y.; Takada, T.; Suzuki, H. Novel dysfunctional variant in ABCG2 as a cause of severe tophaceous gout: Biochemical, molecular genetics and functional analysis. Rheumatology 2016, 55, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Pavelcova, K.; Zavada, J.; Petru, L.; Simek, P.; Cepek, P.; Pavlikova, M.; Matsuo, H.; Merriman, T.R.; Pavelka, K. Functional non-synonymous variants of ABCG2 and gout risk. Rheumatology 2017, 56, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Higashino, T.; Takada, T.; Nakaoka, H.; Toyoda, Y.; Stiburkova, B.; Miyata, H.; Ikebuchi, Y.; Nakashima, H.; Shimizu, S.; Kawaguchi, M.; et al. Multiple common and rare variants of ABCG2 cause gout. RMD Open 2017, 3, e000464. [Google Scholar] [CrossRef]
- Woodward, O.M.; Tukaye, D.N.; Cui, J.; Greenwell, P.; Constantoulakis, L.M.; Parker, B.S.; Rao, A.; Kottgen, M.; Maloney, P.C.; Guggino, W.B. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 5223–5228. [Google Scholar] [CrossRef]
- Mizuarai, S.; Aozasa, N.; Kotani, H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int. J. Cancer 2004, 109, 238–246. [Google Scholar] [CrossRef]
- Cleophas, M.C.; Joosten, L.A.; Stamp, L.K.; Dalbeth, N.; Woodward, O.M.; Merriman, T.R. ABCG2 polymorphisms in gout: Insights into disease susceptibility and treatment approaches. Pharmgenom. Pers. Med. 2017, 10, 129–142. [Google Scholar] [CrossRef]
- Dehghan, A.; Köttgen, A.; Yang, Q.; Hwang, S.J.; Kao, W.L.; Rivadeneira, F.; Boerwinkle, E.; Levy, D.; Hofman, A.; Astor, B.C.; et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet 2008, 372, 1953–1961. [Google Scholar] [CrossRef]
- Li, R.; Miao, L.; Qin, L.; Xiang, Y.; Zhang, X.; Peng, H.; Mailamuguli; Sun, Y.; Yao, H. A meta-analysis of the associations between the Q141K and Q126X ABCG2 gene variants and gout risk. Int. J. Clin. Exp. Pathol. 2015, 8, 9812–9823. [Google Scholar]
- Roberts, R.L.; Wallace, M.C.; Phipps-Green, A.J.; Topless, R.; Drake, J.M.; Tan, P.; Dalbeth, N.; Merriman, T.R.; Stamp, L.K. ABCG2 loss-of-function polymorphism predicts poor response to allopurinol in patients with gout. Pharm. J. 2017, 17, 201–203. [Google Scholar] [CrossRef]
- Mosaffa, F.; Lage, H.; Afshari, J.T.; Behravan, J. Interleukin-1 beta and tumor necrosis factor-alpha increase ABCG2 expression in MCF-7 breast carcinoma cell line and its mitoxantrone-resistant derivative, MCF-7/MX. Inflamm. Res. 2009, 58, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Evseenko, D.A.; Paxton, J.W.; Keelan, J.A. Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab. Dispos. 2007, 35, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, M.; Bembinster, L.A.; Baumgarten, S.C.; Frasor, J. Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NFκB cooperativity at adjacent response elements. J. Biol. Chem. 2010, 285, 31100–31106. [Google Scholar] [CrossRef] [PubMed]
- Stiburkova, B.; Pavelcova, K.; Pavlikova, M.; Ješina, P.; Pavelka, K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res. Ther. 2019, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Mančíková, A.; Krylov, V.; Morimoto, K.; Pavelcová, K.; Bohatá, J.; Pavelka, K.; Pavlíková, M.; Suzuki, H.; Matsuo, H.; et al. Functional characterization of clinically-relevant rare variants in ABCG2 identified in a gout and hyperuricemia cohort. Cells 2019, 8, 363. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.L.; Robinson, H.; Masi, A.T.; Decker, J.L.; McCarty, D.J.; Yü, T.F. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977, 20, 895–900. [Google Scholar] [CrossRef]
- Takada, T.; Ichida, K.; Matsuo, H.; Nakayama, A.; Murakami, K.; Yamanashi, Y.; Kasuga, H.; Shinomiya, N.; Suzuki, H. ABCG2 dysfunction increases serum uric acid by decreased intestinal urate excretion. Nucleosides Nucleotides Nucleic Acids 2014, 33, 275–281. [Google Scholar] [CrossRef]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Nakaoka, H.; Nakamura, T.; Nakashima, H.; Takada, Y.; Oikawa, Y.; Takada, T.; Sakiyama, M.; Shimizu, S.; et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Sci. Rep. 2014, 4, 5227. [Google Scholar] [CrossRef]
- Aune, D.; Norat, T.; Vatten, L.J. Body mass index and the risk of gout: A systematic review and dose–response meta-analysis of prospective studies. Eur. J. Nutr. 2014, 53, 1591–1601. [Google Scholar] [CrossRef]
- Evrin, P.-E.; Nilsson, S.E.; Öberg, T.; Malmberg, B. Serum C-reactive protein in elderly men and women: Association with mortality, morbidity and various biochemical values. Scand. J. Clin. Lab. Investig. 2005, 65, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Wyczalkowska-Tomasik, A.; Czarkowska-Paczek, B.; Zielenkiewicz, M.; Paczek, L. Inflammatory markers change with age, but do not fall beyond reported normal ranges. Arch. Immunol. Ther. Exp. 2016, 64, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, E. Reduced glomerular function and prevalence of gout: NHANES 2009–10. PLoS ONE 2012, 7, e50046. [Google Scholar] [CrossRef]
- Rothenbacher, D.; Primatesta, P.; Ferreira, A.; Cea-Soriano, L.; Rodriguez, L.A.G. Frequency and risk factors of gout flares in a large population-based cohort of incident gout. Rheumatology 2011, 50, 973–981. [Google Scholar] [CrossRef]
- Wang, X.; Wu, X.; Wang, C.; Zhang, W.; Ouyang, Y.; Yu, Y.; He, Z. Transcriptional suppression of breast cancer resistance protein (BCRP) by wild-type p53 through the NF-κB pathway in MCF-7 cells. FEBS Lett. 2010, 584, 3392–3397. [Google Scholar] [CrossRef]
- Shen, S.; Callaghan, D.; Juzwik, C.; Xiong, H.; Huang, P.; Zhang, W. ABCG2 reduces ROS-mediated toxicity and inflammation: A potential role in Alzheimer’s disease. J. Neurochem. 2010, 114, 1590–1604. [Google Scholar] [CrossRef]
- Von Wedel-Parlow, M.; Wölte, P.; Galla, H.-J. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. J. Neurochem. 2009, 111, 111–118. [Google Scholar] [CrossRef]
- Mosaffa, F.; Kalalinia, F.; Lage, H.; Afshari, J.T.; Behravan, J. Pro-inflammatory cytokines interleukin-1 beta, interleukin 6, and tumor necrosis factor-alpha alter the expression and function of ABCG2 in cervix and gastric cancer cells. Mol. Cell. Biochem. 2012, 363, 385–393. [Google Scholar] [CrossRef]
- Deuring, J.J.; de Haar, C.; Koelewijn, C.L.; Kuipers, E.J.; Peppelenbosch, M.P.; van der Woude, C.J. Absence of ABCG2-mediated mucosal detoxification in patients with active inflammatory bowel disease is due to impeded protein folding. Biochem. J. 2012, 441, 87–93. [Google Scholar] [CrossRef]
- Wen, T.; Rothenberg, M.E. The regulatory function of eosinophils. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Kienhorst, L.B.E.; van Lochem, E.; Kievit, W.; Dalbeth, N.; Merriman, M.E.; Phipps-Green, A.; Loof, A.; van Heerde, W.; Vermeulen, S.; Stamp, L.K.; et al. Gout is a chronic inflammatory disease in which high levels of interleukin-8 (CXCL8), myeloid-related protein 8/myeloid-related protein 14 complex, and an altered proteome are associated with diabetes mellitus and cardiovascular disease. Arthritis Rheumatol. 2015, 67, 3303–3313. [Google Scholar] [CrossRef]
- Estevez-Garcia, I.O.; Gallegos-Nava, S.; Vera-Pérez, E.; Silveira, L.H.; Ventura-Ríos, L.; Vancini, G.; Hernández-Díaz, C.; Sánchez-Muñoz, F.; Ballinas-Verdugo, M.A.; Gutierrez, M.; et al. Levels of cytokines and microRNAs in individuals with asymptomatic hyperuricemia and ultrasonographic findings of gout: A bench-to-bedside approach. Arthritis Care Res. 2018, 70, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg-Hasson, Y.; Hansmann, L.; Liedtke, M.; Herschmann, I.; Maecker, H.T. Effects of serum and plasma matrices on multiplex immunoassays. Immunol. Res. 2014, 58, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, A.R.; Wu, H.; Xydakis, A.M.; Jones, P.H.; Smith, E.O.; Sweeney, J.F.; Corry, D.B.; Ballantyne, C.M. Eotaxin and obesity. J. Clin. Endocrinol. Metab. 2006, 91, 256–261. [Google Scholar] [CrossRef]
- Grainger, R.; McLaughlin, R.J.; Harrison, A.A.; Harper, J.L. Hyperuricaemia elevates circulating CCL2 levels and primes monocyte trafficking in subjects with inter-critical gout. Rheumatology 2013, 52, 1018–1021. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, L.; Jiang, L.; Wen, P.; Cao, H.; He, W.; Dai, C.; Yang, J. Uric acid induces renal inflammation via activating tubular NF-κB signaling pathway. PLoS ONE 2012, 7, e39738. [Google Scholar] [CrossRef]
- Mcnearney, T.; Baethge, B.A.; Cao, S.; Alam, R.; Lisse, J.R.; Westlund, K.N. Excitatory amino acids, TNF-alpha, and chemokine levels in synovial fluids of patients with active arthropathies. Clin. Exp. Immunol. 2004, 137, 621–627. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).