A Personalized CYP2C19 Phenotype-Guided Dosing Regimen of Voriconazole Using a Population Pharmacokinetic Analysis

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Study Population
- Study 1: a single dose of intravenous voriconazole 200 mg, followed by single and multiple doses of oral voriconazole 200 mg every 12 h [9],
- Study 2: a single dose of oral voriconazole 400 mg [21],
- Study 3: a single dose of intravenous voriconazole 200 mg [20],
- Study 4: a single dose of intravenous voriconazole 200 mg, followed by a single dose of oral voriconazole 200 mg [22],
- Study 5: loading dose of intravenous voriconazole 6 mg/kg or oral voriconazole 400 mg every 12 h on the first day, followed by TDM-based maintenance doses of intravenous voriconazole 4 mg/kg or oral voriconazole 200 mg every 12 h [16].
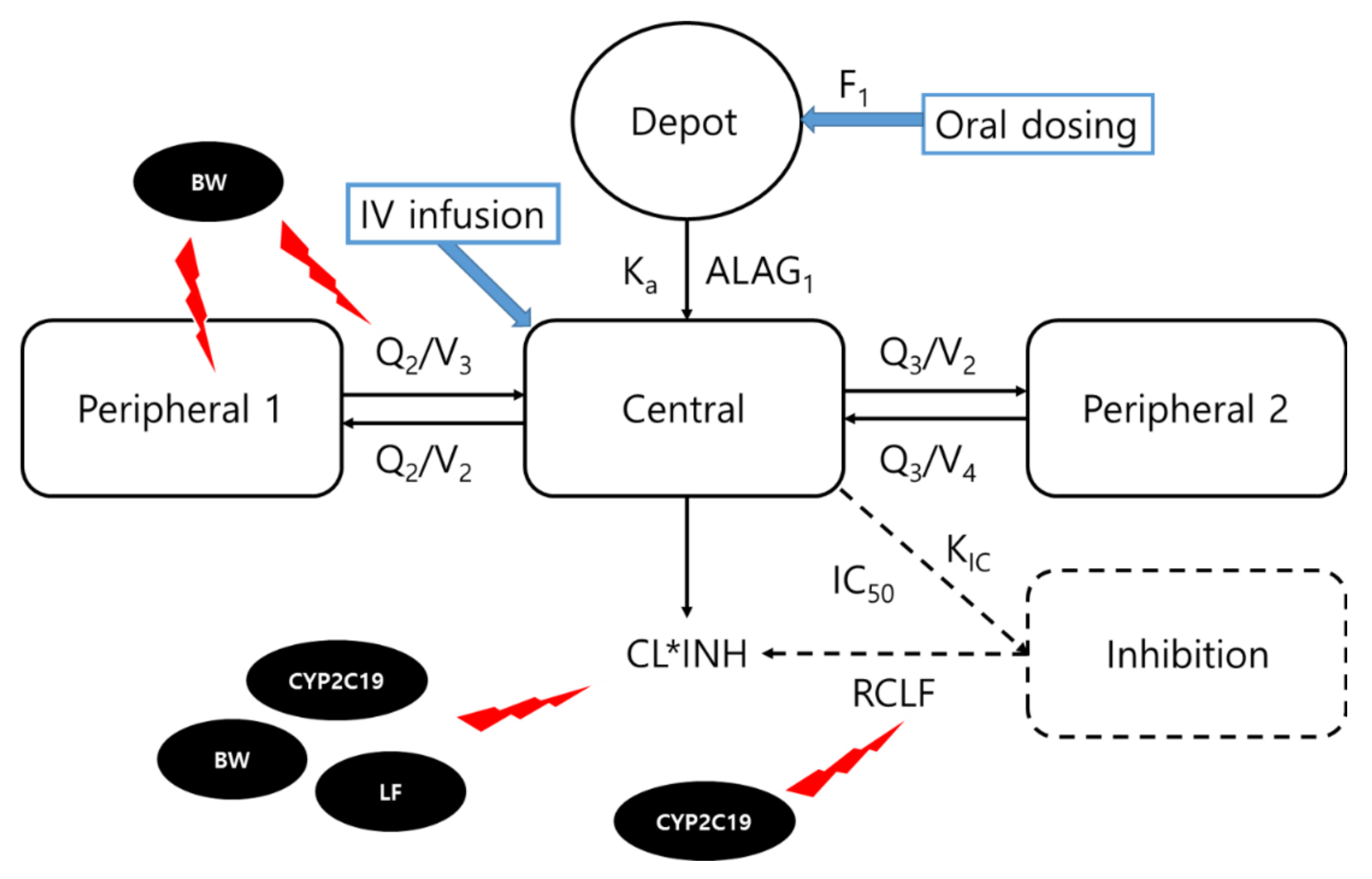
2.2. Population Pharmacokinetic Analysis
2.3. Model Selection and Validation
2.4. Model-Based Simulation
3. Results
3.1. Demographics
3.2. Population Pharmacokinetic Model
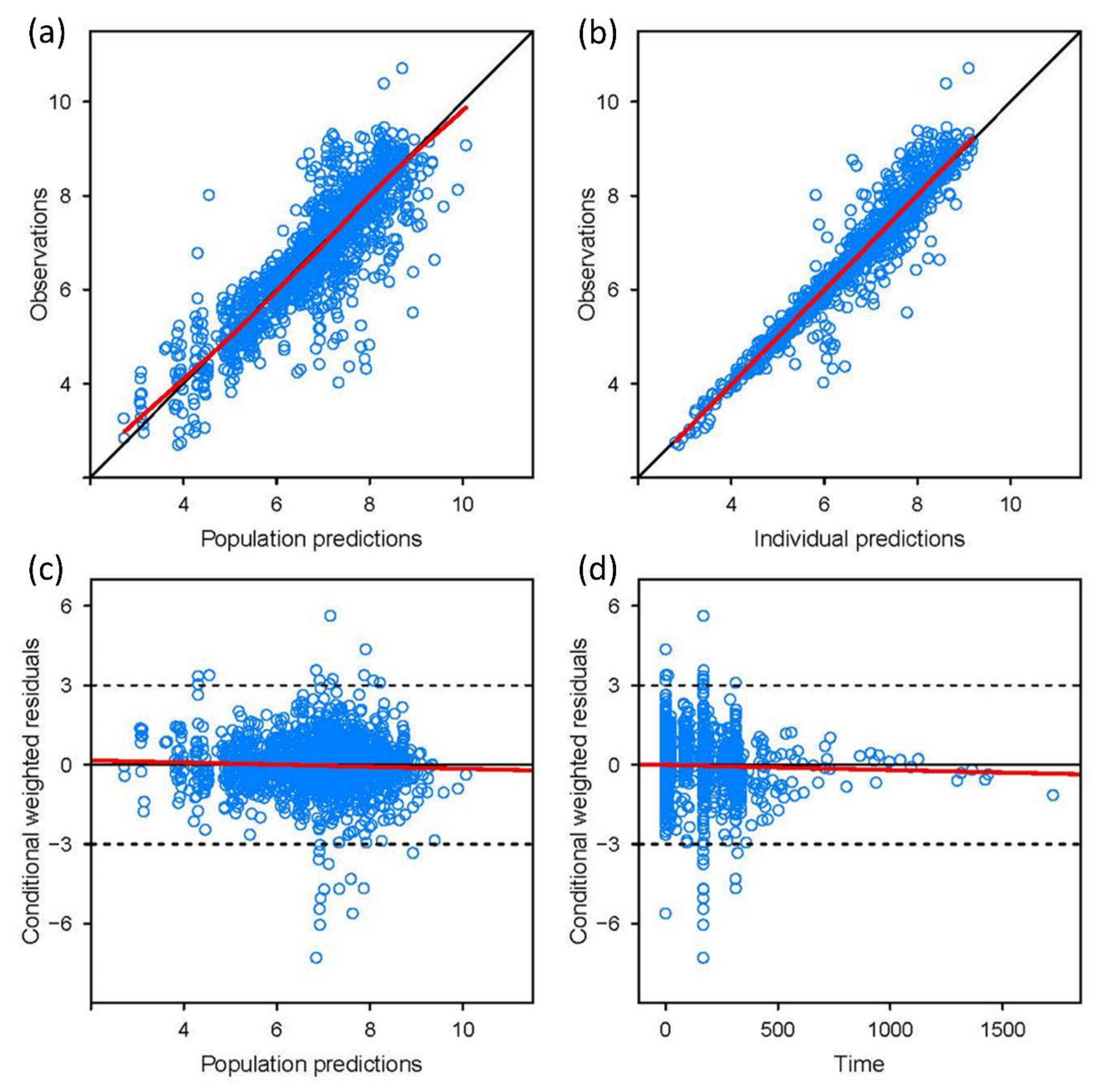
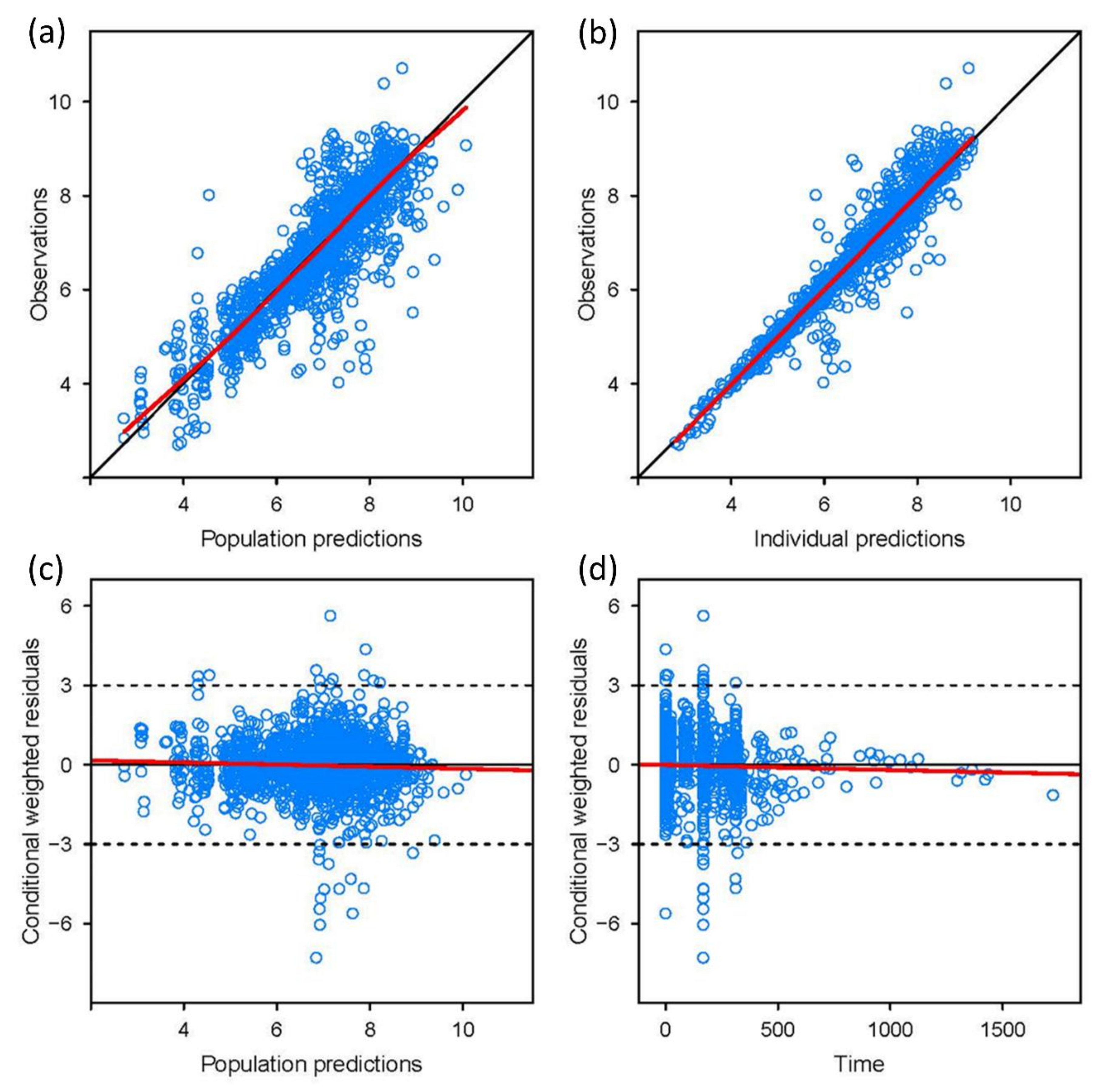
3.3. Model Validation
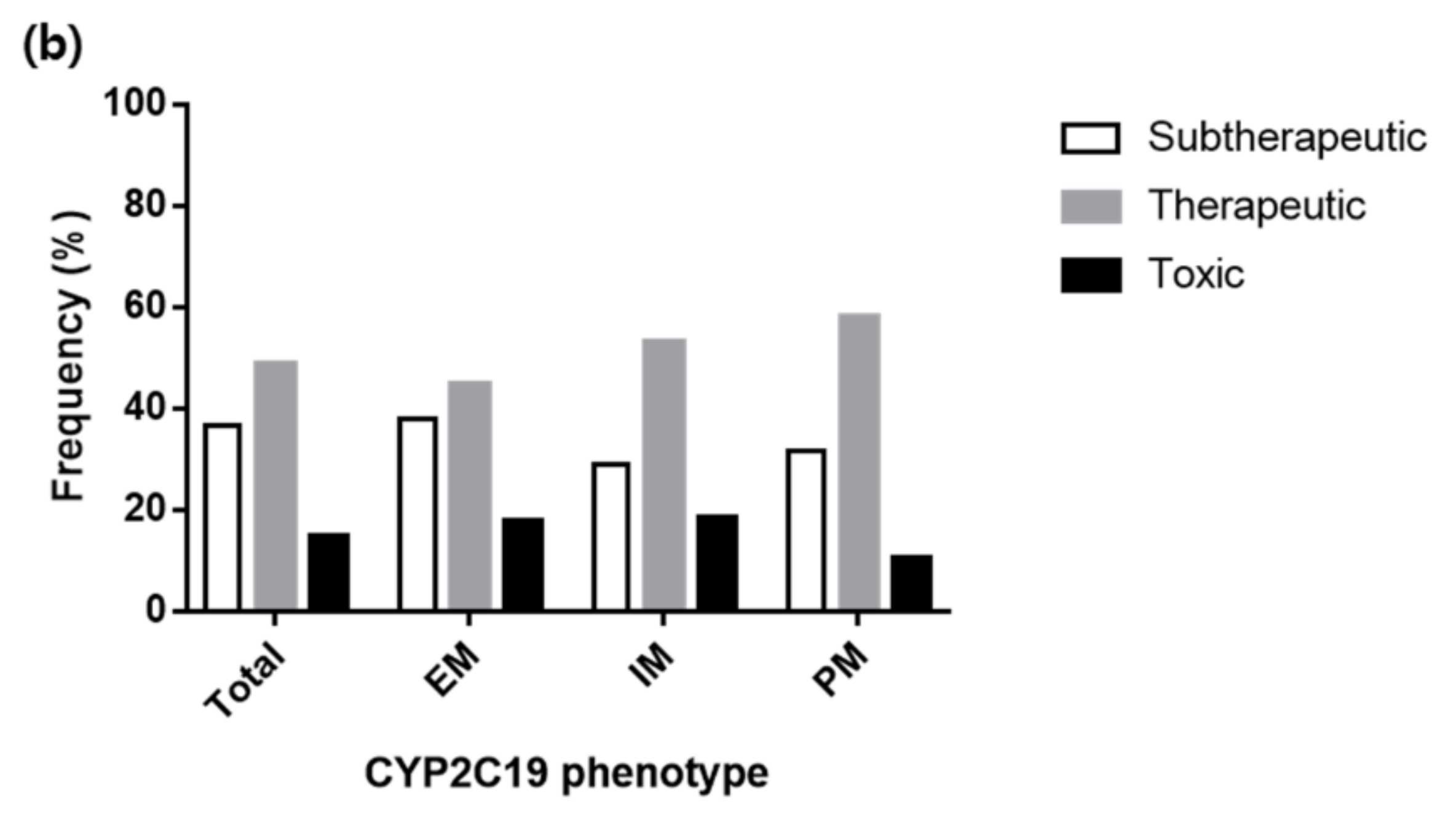
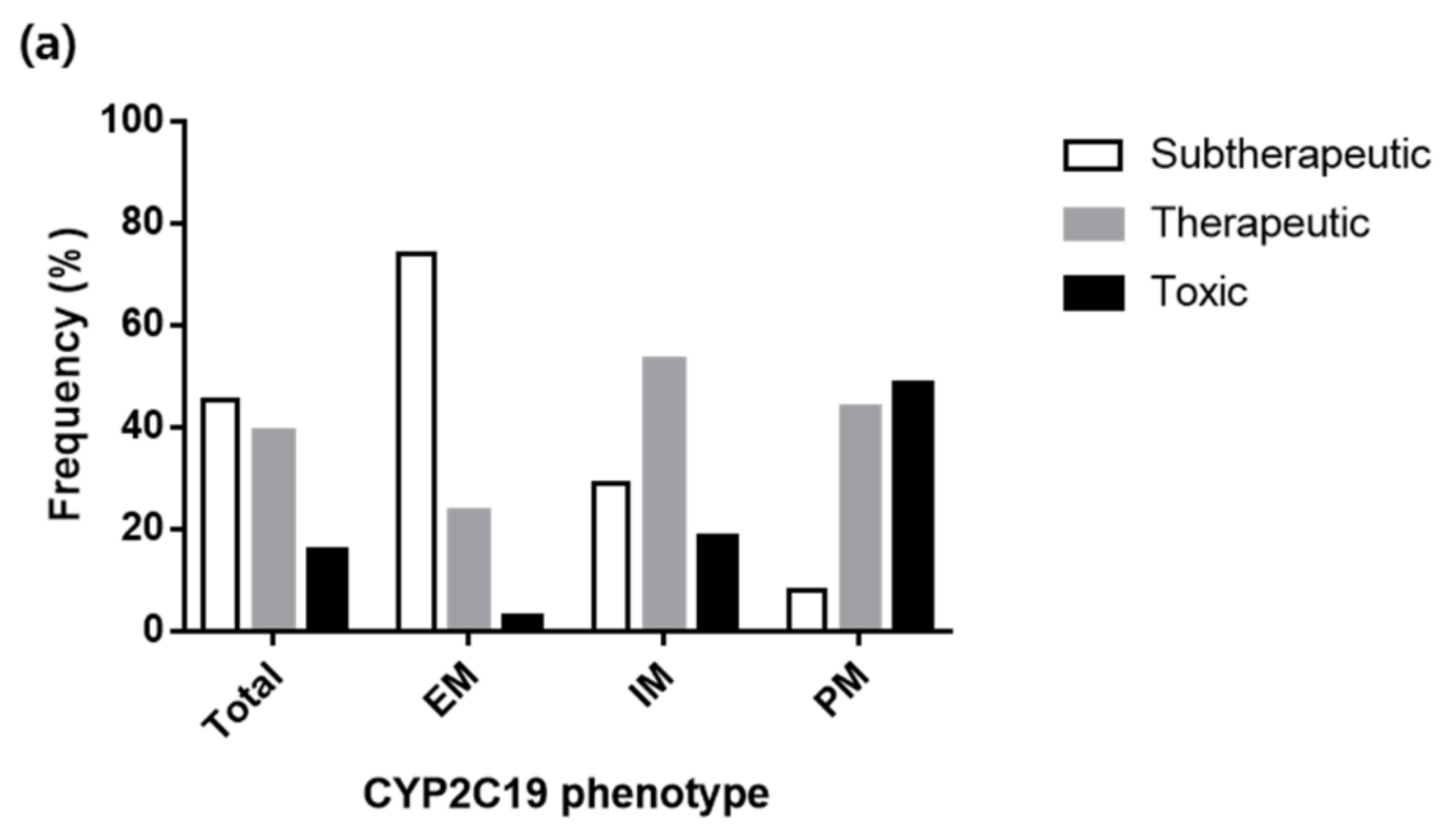
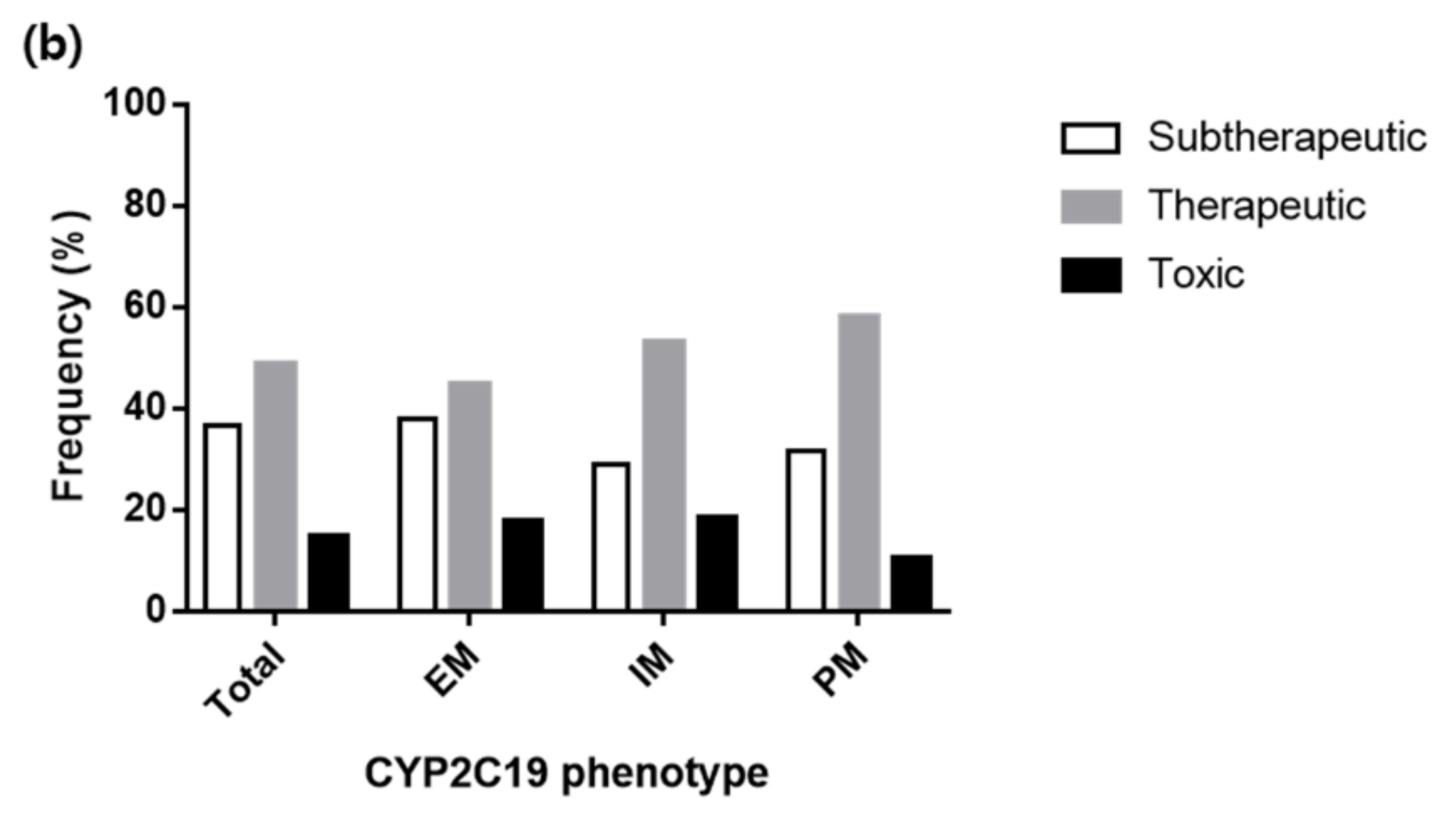
3.4. Various Dosing Regimens According to the CYP2C19 Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Johnson, L.B.; Kauffman, C.A. Voriconazole: A new triazole antifungal agent. Clin. Infect. Dis. 2003, 36, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Yu, C.Y. Voriconazole against fluconazole-susceptible and resistant candida isolates: In-vitro efficacy compared with that of itraconazole and ketoconazole. J. Antimicrob. Chemother. 1998, 42, 253–256. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Ihle, F.; Derendorf, H. Pharmacokinetic/pharmacodynamic profile of voriconazole. Clin. Pharmacokinet. 2006, 45, 649–663. [Google Scholar] [CrossRef]
- Imhof, A.; Schaer, D.J.; Schanz, U.; Schwarz, U. Neurological adverse events to voriconazole: Evidence for therapeutic drug monitoring. Swiss Med. Wkly. 2006, 136, 739–742. [Google Scholar] [CrossRef]
- Denning, D.W.; Ribaud, P.; Milpied, N.; Caillot, D.; Herbrecht, R.; Thiel, E.; Haas, A.; Ruhnke, M.; Lode, H. Efficacy and safety of voriconazole in the treatment of acute invasive aspergillosis. Clin. Infect. Dis. 2002, 34, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Brayshaw, N.; Tomaszewski, K.; Troke, P.; Wood, N. Investigation of the potential relationships between plasma voriconazole concentrations and visual adverse events or liver function test abnormalities. J. Clin. Pharmacol. 2006, 46, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Purkins, L.; Wood, N.; Ghahramani, P.; Greenhalgh, K.; Allen, M.J.; Kleinermans, D. Pharmacokinetics and safety of voriconazole following intravenous- to oral-dose escalation regimens. Antimicrob. Agents Chemother. 2002, 46, 2546–2553. [Google Scholar] [CrossRef]
- Scholz, I.; Oberwittler, H.; Riedel, K.D.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br. J. Clin. Pharmacol. 2009, 68, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, B.H.; Nam, W.S.; Yoon, S.H.; Cho, J.Y.; Shin, S.G.; Jang, I.J.; Yu, K.S. Effect of CYP2C19 polymorphism on the pharmacokinetics of voriconazole after single and multiple doses in healthy volunteers. J. Clin. Pharmacol. 2012, 52, 195–203. [Google Scholar] [CrossRef]
- Jeong, S.; Nguyen, P.D.; Desta, Z. Comprehensive in vitro analysis of voriconazole inhibition of eight cytochrome P450 (CYP) enzymes: Major effect on CYPs 2B6, 2C9, 2C19, and 3A. Antimicrob. Agents Chemother. 2009, 53, 541–551. [Google Scholar] [CrossRef]
- Hohmann, N.; Kreuter, R.; Blank, A.; Weiss, J.; Burhenne, J.; Haefeli, W.E.; Mikus, G. Autoinhibitory properties of the parent but not of the N-oxide metabolite contribute to infusion rate-dependent voriconazole pharmacokinetics. Br. J. Clin. Pharmacol. 2017, 83, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, N.; Kocheise, F.; Carls, A.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Dose-Dependent Bioavailability and CYP3A Inhibition Contribute to Non-Linear Pharmacokinetics of Voriconazole. Clin. Pharmacokinet. 2016, 55, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Pascual, A.; Calandra, T.; Bolay, S.; Buclin, T.; Bille, J.; Marchetti, O. Voriconazole therapeutic drug monitoring in patients with invasive mycoses improves efficacy and safety outcomes. Clin. Infect. Dis. 2008, 46, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Owusu Obeng, A.; Egelund, E.F.; Alsultan, A.; Peloquin, C.A.; Johnson, J.A. CYP2C19 polymorphisms and therapeutic drug monitoring of voriconazole: Are we ready for clinical implementation of pharmacogenomics? Pharmacotherapy 2014, 34, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Safdar, N.; Knasinski, V.; Simmons, W.; Bhavnani, S.M.; Ambrose, P.G.; Andes, D. Voriconazole therapeutic drug monitoring. Antimicrob. Agents Chemother. 2006, 50, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Park, W.B.; Kim, N.H.; Kim, K.H.; Lee, S.H.; Nam, W.S.; Yoon, S.H.; Song, K.H.; Choe, P.G.; Kim, N.J.; Jang, I.J.; et al. The effect of therapeutic drug monitoring on safety and efficacy of voriconazole in invasive fungal infections: A randomized controlled trial. Clin. Infect. Dis. 2012, 55, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Teusink, A.; Vinks, A.; Zhang, K.; Davies, S.; Fukuda, T.; Lane, A.; Nortman, S.; Kissell, D.; Dell, S.; Filipovich, A.; et al. Genotype-Directed Dosing Leads to Optimized Voriconazole Levels in Pediatric Patients Receiving Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 482–486. [Google Scholar] [CrossRef]
- Lin, X.B.; Li, Z.W.; Yan, M.; Zhang, B.K.; Liang, W.; Wang, F.; Xu, P.; Xiang, D.X.; Xie, X.B.; Yu, S.J.; et al. Population pharmacokinetics of voriconazole and CYP2C19 polymorphisms for optimizing dosing regimens in renal transplant recipients. Br. J. Clin. Pharmacol. 2018, 84, 1587–1597. [Google Scholar] [CrossRef]
- Moriyama, B.; Obeng, A.O.; Barbarino, J.; Penzak, S.R.; Henning, S.A.; Scott, S.A.; Agúndez, J.A.; Wingard, J.R.; McLeod, H.L.; Klein, T.E.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP2C19 and Voriconazole Therapy. Clin. Pharmacol. Ther. 2018, 103, 349. [Google Scholar] [CrossRef]
- Chung, H.; Lee, H.; Han, H.K.; An, H.; Lim, K.S.; Lee, Y.J.; Cho, J.Y.; Yoon, S.H.; Jang, I.J.; Yu, K.S. A pharmacokinetic comparison of two voriconazole formulations and the effect of CYP2C19 polymorphism on their pharmacokinetic profiles. Drug Des. Dev. Ther. 2015, 9, 2609–2616. [Google Scholar] [CrossRef]
- Lee, S. Inductive effect of rifampin on the clinical pharmacokinetics of voriconazole and related biomarkers. Ph.D. Thesis, Seoul National University College of Medicine, Seoul, Korea, 2012. [Google Scholar]
- Lee, S.W. Oral absorption of voriconazole is affected by SLCO2B1 c.*396T>C genetic polymorphism in CYP2C19 poor metabolizers. Unpublished work. 2019. [Google Scholar]
- Plock, N.; Buerger, C.; Joukhadar, C.; Kljucar, S.; Kloft, C. Does linezolid inhibit its own metabolism? Population pharmacokinetics as a tool to explain the observed nonlinearity in both healthy volunteers and septic patients. Drug Metab. Dispos. 2007, 35, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Institute, N.C. Common Terminology Criteria for Adverse Events (CTCAE); U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute: Bethesda, MD, USA, 2010. [Google Scholar]
- Sheiner, L.B.; Beal, S.L. Some suggestions for measuring predictive performance. J. Pharmacokinet. Biopharm. 1981, 9, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Pascual, A.; Csajka, C.; Buclin, T.; Bolay, S.; Bille, J.; Calandra, T.; Marchetti, O. Challenging recommended oral and intravenous voriconazole doses for improved efficacy and safety: Population pharmacokinetics-based analysis of adult patients with invasive fungal infections. Clin. Infect. Dis. 2012, 55, 381–390. [Google Scholar] [CrossRef]
- Wang, T.; Chen, S.; Sun, J.; Cai, J.; Cheng, X.; Dong, H.; Wang, X.; Xing, J.; Dong, W.; Yao, H.; et al. Identification of factors influencing the pharmacokinetics of voriconazole and the optimization of dosage regimens based on Monte Carlo simulation in patients with invasive fungal infections. J. Antimicrob. Chemother. 2014, 69, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Hope, W.W. Population pharmacokinetics of voriconazole in adults. Antimicrob. Agents Chemother. 2012, 56, 526–531. [Google Scholar] [CrossRef]
- Dolton, M.J.; Mikus, G.; Weiss, J.; Ray, J.E.; McLachlan, A.J. Understanding variability with voriconazole using a population pharmacokinetic approach: Implications for optimal dosing. J. Antimicrob. Chemother. 2014, 69, 1633–1641. [Google Scholar] [CrossRef]
- Karlsson, M.O.; Lutsar, I.; Milligan, P.A. Population pharmacokinetic analysis of voriconazole plasma concentration data from pediatric studies. Antimicrob. Agents Chemother. 2009, 53, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Friberg, L.E.; Ravva, P.; Karlsson, M.O.; Liu, P. Integrated population pharmacokinetic analysis of voriconazole in children, adolescents, and adults. Antimicrob. Agents Chemother. 2012, 56, 3032–3042. [Google Scholar] [CrossRef] [PubMed]
- McDougall, D.A.; Martin, J.; Playford, E.G.; Green, B. Determination of a suitable voriconazole pharmacokinetic model for personalised dosing. J. Pharmacokinet. Pharmacodyn. 2016, 43, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Inc. VFEND (Voriconazole) [Prescribing Information]. 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021266s038,021267s047,021630s028lbl.pdf (accessed on 9 February 2019).
- Qi, F.; Zhu, L.; Li, N.; Ge, T.; Xu, G.; Liao, S. Influence of different proton pump inhibitors on the pharmacokinetics of voriconazole. Int. J. Antimicrob. Agents 2017, 49, 403–409. [Google Scholar] [CrossRef]
- Yasu, T.; Konuma, T.; Kato, S.; Kurokawa, Y.; Takahashi, S.; Tojo, A. Different effects of lansoprazole and rabeprazole on the plasma voriconazole trough levels in allogeneic hematopoietic cell transplant recipients. Ann. Hematol. 2016, 95, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Peng, F.H.; Yan, M.; Liang, W.; Liu, X.L.; Wu, Y.Q.; Lin, X.B.; Tan, S.L.; Wang, F.; Xu, P.; et al. Impact of CYP2C19 Genotype and Liver Function on Voriconazole Pharmacokinetics in Renal Transplant Recipients. Ther. Drug Monit. 2017, 39, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Y.; Liu, W.; Chen, K.; Liang, S.Y.; Liu, F. The influence of combination use of CYP450 inducers on the pharmacokinetics of voriconazole: A systematic review. J. Clin. Pharm. Ther. 2017, 42, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Dolton, M.J.; Ray, J.E.; Chen, S.C.; Ng, K.; Pont, L.G.; McLachlan, A.J. Multicenter study of voriconazole pharmacokinetics and therapeutic drug monitoring. Antimicrob. Agents Chemother. 2012, 56, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Radford, S.A.; Johnson, E.M.; Warnock, D.W. In vitro studies of activity of voriconazole (UK-109,496), a new triazole antifungal agent, against emerging and less-common mold pathogens. Antimicrob. Agents Chemother. 1997, 41, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Mangal, N.; Hamadeh, I.S.; Arwood, M.J.; Cavallari, L.H.; Samant, T.S.; Klinker, K.P.; Bulitta, J.; Schmidt, S. Optimization of Voriconazole Therapy for the Treatment of Invasive Fungal Infections in Adults. Clin. Pharmacol. Ther. 2018, 104, 957–965. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total (n = 193) | Healthy Subjects a (n = 93) | Patients b (n = 100) |
|---|---|---|---|
| Age (years) | 34 (18–80) | 26 (20–41) | 59 (18–80) |
| Weight (kg) | 66.0 (40.8–88.5) | 70.3 (57.6–88.5) | 59.4 (40.8–86.4) |
| Aspartate aminotransferase (U/L) | 21 (7–377) | 18 (9–40) | 30 (7–377) |
| Alanine aminotransferase (U/L) | 21 (4–363) | 15 (4–52) | 29 (4–363) |
| Sex | |||
| Male | 164 (85) | 93 (100) | 71 (71) |
| Female | 29 (15) | - | 29 (29) |
| CYP2C19 phenotype | |||
| Extensive metabolizer | 75 (39) | 32 (34) | 43 (43) |
| Intermediate metabolizer | 70 (36) | 27 (29) | 43 (43) |
| Poor metabolizer | 48 (25) | 34 (37) | 14 (14) |
| Liver function abnormality c | |||
| Grade 0 | 165 (85.5) | 93 (100) | 72 (72) |
| Grade 1 | 9 (4.7) | - | 9 (9) |
| Grade 2 | 13 (6.7) | - | 13 (13) |
| Grade 3 | 5 (2.6) | - | 5 (5) |
| Grade 4 | 1 (0.5) | - | 1 (1) |
| Co-medication | |||
| Proton pump inhibitors | 22 (11.4) | - | 22 (22) |
| Steroids | 9 (4.7) | - | 9 (9) |
| Parameters | Estimates | RSE (%) |
|---|---|---|
| Structural model | ||
| V2; central volume of distribution (L) | 35.7 | 15.7 |
| CL; clearance (L/h) | 45.3 | 5.8 |
| V3; peripheral 1 volume of distribution (L) | 58.9 | 6.2 |
| Q2; inter-compartmental clearance between central and peripheral 1 compartment (L/h) | 10.9 | 8.0 |
| V4; peripheral 2 volume of distribution (L) | 25.4 | 16.7 |
| Q3; inter-compartmental clearance between central and peripheral 2 compartment (L/h) | 54.6 | 45.4 |
| Ka; absorption rate constant (h−1) | 1.23 | 15.4 |
| F1; bioavailability | 0.876 | 2.3 |
| ALAG1; absorption lag-time (h) | 0.237 | 1.8 |
| RCLF; fraction of clearance which cannot be inhibited | 0.162 | 9.7 |
| IC50; concentration in the inhibition compartment yielding 50% of maximum clearance inhibition | 0.01 FIX | NA |
| KIC; rate constant into inhibition compartment | 0.002 | 14.9 |
| Inter-individual variability (IIV) | ||
| IIV for V2 (% CV) | 40.2 | 23.3 a |
| IIV for CL (% CV) | 21.4 | 10.6 a |
| IIV for V3 (% CV) | 20.6 | 34.1 a |
| IIV for Q2 (% CV) | 28.8 | 20.0 a |
| IIV for Ka (% CV) | 87.8 | 14.4 a |
| IIV for F1 (% CV) | 84.4 | 20.3 a |
| IIV for RCLF (% CV) | 54.4 | 13.0 a |
| Correlation between V2 and CL | 0.0116 | 95.7 b |
| Correlation between V2 and V3 | −0.0117 | 200.9 b |
| Correlation between V2 and Q2 | −0.0734 | 49.2 b |
| Correlation between CL and V3 | −0.0119 | 72.5 b |
| Correlation between CL and Q2 | 0.008 | 150.3 b |
| Correlation between V3 and Q2 | 0.0345 | 67.0 b |
| Residual variability | ||
| Additive error for healthy subjects (mg/L) | 0.208 | 8.4 |
| Additive error for patients (mg/L) | 0.799 | 6.7 |
| Variable | Estimates | RSE (%) |
|---|---|---|
| Effect on CL | ||
| Body weight exponent for CL | 0.595 | 31.8 |
| CYP2C19 phenotype effect for CL (cf. 0 for extensive metabolizer) | ||
| Intermediate metabolizer | −0.186 a | 29.5 a |
| Poor metabolizer | −0.746 a | 10.9 a |
| Liver function abnormality effect for CL (cf. 0 for grade < 3) | ||
| Grade ≥ 3 | −0.75 | 49.3 |
| Effect on V3 | ||
| Body weight exponent for V3 | 2.2 | 20.0 |
| Effect on Q2 | ||
| Body weight exponent for Q2 | 2.56 | 18.1 |
| Effect on RCLF | ||
| CYP2C19 phenotype effect for CL (cf. 0 for extensive metabolizer) | ||
| Intermediate metabolizer | −0.51 a | 27.5 a |
| Poor metabolizer | −0.44 a | 42.3 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Rhee, S.-j.; Park, W.B.; Yu, K.-S.; Jang, I.-J.; Lee, S. A Personalized CYP2C19 Phenotype-Guided Dosing Regimen of Voriconazole Using a Population Pharmacokinetic Analysis. J. Clin. Med. 2019, 8, 227. https://doi.org/10.3390/jcm8020227
Kim Y, Rhee S-j, Park WB, Yu K-S, Jang I-J, Lee S. A Personalized CYP2C19 Phenotype-Guided Dosing Regimen of Voriconazole Using a Population Pharmacokinetic Analysis. Journal of Clinical Medicine. 2019; 8(2):227. https://doi.org/10.3390/jcm8020227
Chicago/Turabian StyleKim, Yun, Su-jin Rhee, Wan Beom Park, Kyung-Sang Yu, In-Jin Jang, and SeungHwan Lee. 2019. "A Personalized CYP2C19 Phenotype-Guided Dosing Regimen of Voriconazole Using a Population Pharmacokinetic Analysis" Journal of Clinical Medicine 8, no. 2: 227. https://doi.org/10.3390/jcm8020227
APA StyleKim, Y., Rhee, S.-j., Park, W. B., Yu, K.-S., Jang, I.-J., & Lee, S. (2019). A Personalized CYP2C19 Phenotype-Guided Dosing Regimen of Voriconazole Using a Population Pharmacokinetic Analysis. Journal of Clinical Medicine, 8(2), 227. https://doi.org/10.3390/jcm8020227