Podocyte Injury in Lupus Nephritis

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Podocyte Injury in Immune-Complex Lupus Nephritis
3. Proliferative Lupus Nephritis (LN) with Crescent Formation
4. Podocytes as Immune Cells
5. Podocyte Genetics in Lupus Nephritis
6. Lupus Podocytopathy: A Specific Form of LN
7. Conclusions
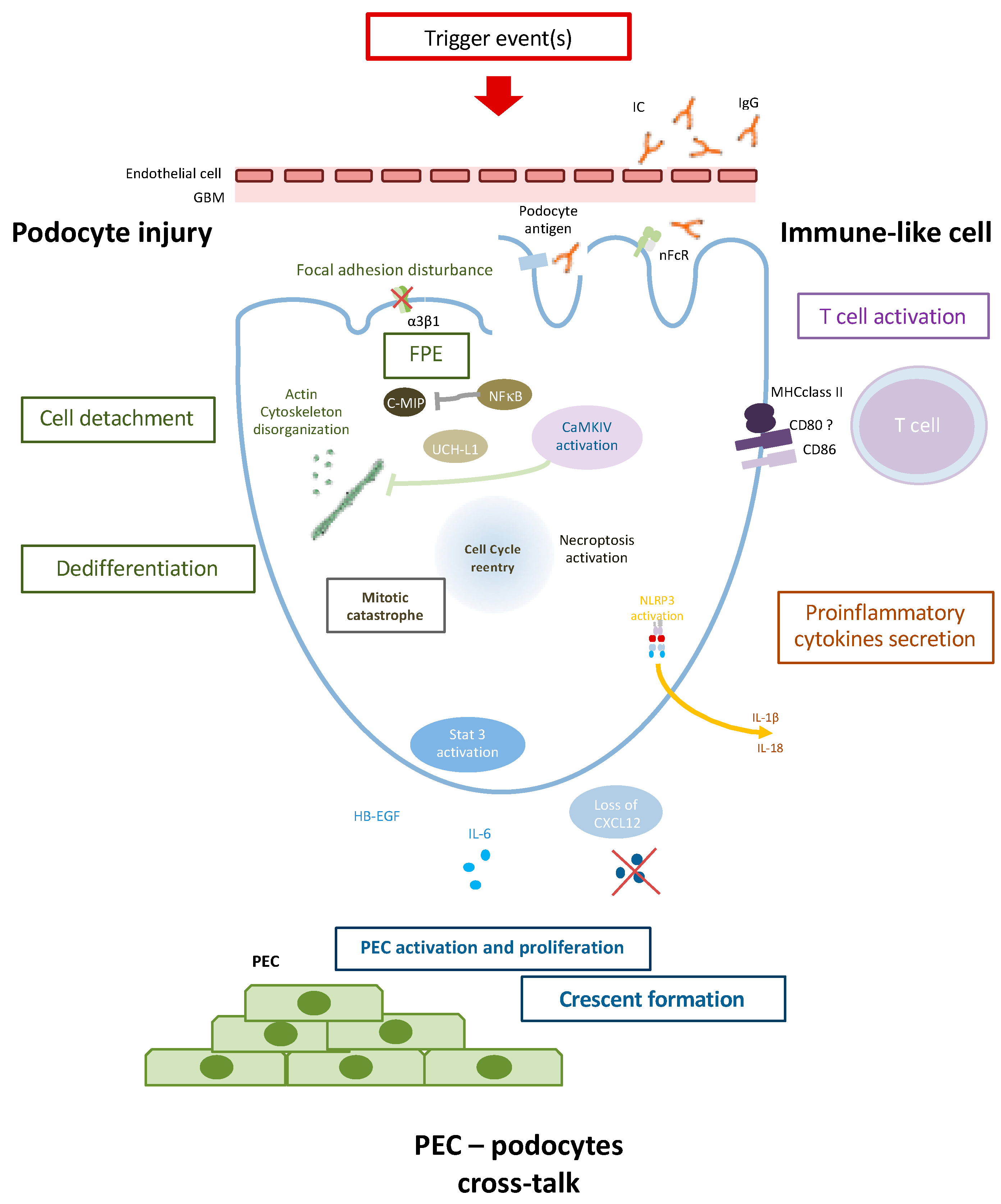
- Podocyte injury is characterized by FPE, loss of podocyte-specific markers, and cell detachment. Actin cytoskeleton disorganisation plays a major in FPE and cell death through mitotic catastrophe. UCH-L1 could as well contribute to podocyte injury by modulating specific podocyte protein expression.
- Podocytes could contribute to the inflammatory process as an APC (antigen-presenting cell). Immunoglobulin (Ig) internalization through neonatal Fc receptors (nFcR) leads to CaMKIV activation and CD86 expression. CD80 and MHC may also be expressed on podocytes during inflammatory process and could contribute to T cell activation. NLRP3 inflammasome activation in the RIP3-dependent pathway could lead to proinflammatory cytokine secretion such as IL-1β and IL-18.
- Podocyte injury finally triggers PEC (Parietal epithelial cell) activation and proliferation through, notably, the Jak/Stat pathway, HB-EGF, and IL-6 production and/or loss of CXCL12 secretion leading to PEC activation and crescent formation.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Tektonidou, M.G.; Dasgupta, A.; Ward, M.M. Risk of End-Stage Renal Disease in Patients With Lupus Nephritis, 1971-2015: A Systematic Review and Bayesian Meta-Analysis. Arthritis Rheumatol. Hoboken NJ 2016, 68, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Weening, J.J.; D’Agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Balow, J.E.; Bruijn, J.A.; Cook, T.; Ferrario, F.; et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J. Am. Soc. Nephrol. JASN 2004, 15, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Bomback, A.S.; Markowitz, G.S. Lupus Podocytopathy: A Distinct Entity. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 547–548. [Google Scholar] [CrossRef]
- Davidson, A. What is damaging the kidney in lupus nephritis? Nat. Rev. Rheumatol. 2016, 12, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.-M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks That Stratify Lupus Patients. Cell 2016, 165, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Haas, M.; Glassock, R.; Zhao, M.-H. Redefining lupus nephritis: Clinical implications of pathophysiologic subtypes. Nat. Rev. Nephrol. 2017, 13, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Sever, S.; Schiffer, M. Actin dynamics at focal adhesions: A common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte-actin dynamics in health and disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef]
- Kriz, W.; Lemley, K.V. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017, 91, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Fries, J.W.; Mendrick, D.L.; Rennke, H.G. Determinants of immune complex-mediated glomerulonephritis. Kidney Int. 1988, 34, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangaku, M.; Shankland, S.J.; Couser, W.G. Cellular response to injury in membranous nephropathy. J. Am. Soc. Nephrol. JASN 2005, 16, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, F.; Song, D.; Wang, S.-X.; Zhao, M.-H. Podocyte involvement in lupus nephritis based on the 2003 ISN/RPS system: A large cohort study from a single centre. Rheumatology (Oxf. Engl.) 2014, 53, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.; Cimbaluk, D.; Lewis, E.J.; Whittier, W.L. Proteinuria in membranous lupus nephritis: The pathology is in the podocyte. Lupus 2013, 22, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Rezende, G.M.; Viana, V.S.; Malheiros, D.M.A.C.; Borba, E.F.; Silva, N.A.S.; Silva, C.; Leon, E.P.; Noronha, I.L.; Bonfa, E. Podocyte injury in pure membranous and proliferative lupus nephritis: Distinct underlying mechanisms of proteinuria? Lupus 2014, 23, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Perysinaki, G.S.; Moysiadis, D.K.; Bertsias, G.; Giannopoulou, I.; Kyriacou, K.; Nakopoulou, L.; Boumpas, D.T.; Daphnis, E. Podocyte main slit diaphragm proteins, nephrin and podocin, are affected at early stages of lupus nephritis and correlate with disease histology. Lupus 2011, 20, 781–791. [Google Scholar] [CrossRef]
- Wang, G.; Lai, F.M.-M.; Tam, L.-S.; Li, K.-M.; Lai, K.-B.; Chow, K.-M.; Li, K.-T.P.; Szeto, C.-C. Messenger RNA expression of podocyte-associated molecules in urinary sediment of patients with lupus nephritis. J. Rheumatol. 2007, 34, 2358–2364. [Google Scholar]
- Perez-Hernandez, J.; Olivares, M.D.; Forner, M.J.; Chaves, F.J.; Cortes, R.; Redon, J. Urinary dedifferentiated podocytes as a non-invasive biomarker of lupus nephritis. Nephrol. Dial. Transplant. 2016, 31, 780–789. [Google Scholar] [CrossRef] [Green Version]
- Kalaaji, M.; Mortensen, E.; Jørgensen, L.; Olsen, R.; Rekvig, O.P. Nephritogenic Lupus Antibodies Recognize Glomerular Basement Membrane-Associated Chromatin Fragments Released from Apoptotic Intraglomerular Cells. Am. J. Pathol. 2006, 168, 1779–1792. [Google Scholar] [CrossRef] [Green Version]
- Kalaaji, M.; Fenton, K.A.; Mortensen, E.S.; Olsen, R.; Sturfelt, G.; Alm, P.; Rekvig, O.P. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kidney Int. 2007, 71, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, M.R.; Wang, C.; Marion, T.N. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. 2012, 82, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J. Am. Soc. Nephrol. JASN 2010, 21, 1912–1927. [Google Scholar] [CrossRef] [PubMed]
- Deocharan, B.; Qing, X.; Lichauco, J.; Putterman, C. α-Actinin Is a Cross-Reactive Renal Target for Pathogenic Anti-DNA Antibodies. J. Immunol. 2002, 168, 3072–3078. [Google Scholar] [CrossRef] [PubMed]
- Mason, L.J.; Ravirajan, C.T.; Rahman, A.; Putterman, C.; Isenberg, D.A. Is alpha-actinin a target for pathogenic anti-DNA antibodies in lupus nephritis? Arthritis Rheumatol. 2004, 50, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Sinico, R.A.; Moroni, G.; Pratesi, F.; Migliorini, P.; Galetti, M.; Murtas, C.; Tincani, A.; Madaio, M.; Radice, A.; et al. Glomerular autoimmune multicomponents of human lupus nephritis in vivo: α-enolase and annexin AI. J. Am. Soc. Nephrol. JASN 2014, 25, 2483–2498. [Google Scholar] [CrossRef] [PubMed]
- Mansur, J.B.; Sabino, A.R.P.; Nishida, S.K.; Kirsztajn, G.M. Is there a role for urinary podocyte excretion assessment in lupus nephritis? Ren. Fail. 2016, 38, 643–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessapt, C.; Baradez, M.O.; Hayward, A.; Dei Cas, A.; Thomas, S.M.; Viberti, G.; Gnudi, L. Mechanical forces and TGFbeta1 reduce podocyte adhesion through alpha3beta1 integrin downregulation. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2009, 24, 2645–2655. [Google Scholar]
- Lasagni, L.; Lazzeri, E.; Shankland, S.J.; Anders, H.-J.; Romagnani, P. Podocyte Mitosis—A Catastrophe. Curr. Mol. Med. 2013, 13, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, L.; Ballerini, L.; Angelotti, M.L.; Parente, E.; Sagrinati, C.; Mazzinghi, B.; Peired, A.; Ronconi, E.; Becherucci, F.; Bani, D.; et al. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells Dayt. Ohio 2010, 28, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, M. The molecular requirements for cytokinesis. Science 2005, 307, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Mühldorfer, J.; Pfister, E.; Büttner-Herold, M.; Klewer, M.; Amann, K.; Daniel, C. Bi-nucleation of podocytes is uniformly accompanied by foot processes widening in renal disease. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2018, 33, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, K.; Yanagida-Asanuma, E.; Faul, C.; Tomino, Y.; Kim, K.; Mundel, P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell Biol. 2006, 8, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Liu, Q.; Zheng, Z.; Fan, J.; Peng, W.; Kong, Q.; He, H.; Yang, S.; Chen, W.; Tang, X.; et al. Tacrolimus Protects Podocytes from Injury in Lupus Nephritis Partly by Stabilizing the Cytoskeleton and Inhibiting Podocyte Apoptosis. PLoS ONE 2015, 10, e0132724. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.-M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Otomo, K.; Yoshida, N.; Abu-Asab, M.S.; Ichinose, K.; Nishino, T.; Kono, M.; Ferretti, A.; Bhargava, R.; Maruyama, S.; et al. CaMK4 compromises podocyte function in autoimmune and nonautoimmune kidney disease. J. Clin. Invest. 2018, 128, 3445–3459. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Kamal, M.; Dahan, K.; Pawlak, A.; Ory, V.; Desvaux, D.; Audard, V.; Candelier, M.; BenMohamed, F.; Mohamed, F.B.; et al. C-mip impairs podocyte proximal signaling and induces heavy proteinuria. Sci. Signal. 2010, 3, ra39. [Google Scholar] [CrossRef]
- Bouachi, K.; Moktefi, A.; Zhang, S.-Y.; Oniszczuk, J.; Sendeyo, K.; Remy, P.; Audard, V.; Pawlak, A.; Ollero, M.; Sahali, D. Expression of CMIP in podocytes is restricted to specific classes of lupus nephritis. PLoS ONE 2018, 13, e0207066. [Google Scholar] [CrossRef]
- Sendeyo, K.; Audard, V.; Zhang, S.; Fan, Q.; Bouachi, K.; Ollero, M.; Rucker-Martin, C.; Gouadon, E.; Desvaux, D.; Bridoux, F.; et al. Upregulation of c-mip is closely related to podocyte dysfunction in membranous nephropathy. Kidney Int. 2013, 83, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Izzedine, H.; Mangier, M.; Ory, V.; Zhang, S.-Y.; Sendeyo, K.; Bouachi, K.; Audard, V.; Péchoux, C.; Soria, J.C.; Massard, C.; et al. Expression patterns of RelA and c-mip are associated with different glomerular diseases following anti-VEGF therapy. Kidney Int. 2014, 85, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Brähler, S.; Ising, C.; Hagmann, H.; Rasmus, M.; Hoehne, M.; Kurschat, C.; Kisner, T.; Goebel, H.; Shankland, S.; Addicks, K.; et al. Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am. J. Physiol. Ren. Physiol. 2012, 303, F1473–F1485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, Y.; Hu, R.; Luo, W.; Mao, X.; Zhao, Z.; Chen, Q.; Zhang, Z. The regulation of the UCH-L1 gene by transcription factor NF-κB in podocytes. Cell. Signal. 2013, 25, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zou, L.-X.; Han, Y.-C.; Wu, L.; Chen, T.; Zhu, D.-D.; Hu, P. A20 overexpression exerts protective effects on podocyte injury in lupus nephritis by downregulating UCH-L1. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwesinger, C.; Meyer, T.N.; Münster, S.; Klug, P.; Saleem, M.; Helmchen, U.; Stahl, R.A.K. A new role for the neuronal ubiquitin C-terminal hydrolase-L1 (UCH-L1) in podocyte process formation and podocyte injury in human glomerulopathies. J. Pathol. 2009, 217, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.; Wu, H.; Wang, T.; Gan, H.; Zhang, X.; Liu, Y.; Li, R.; Zhao, Z.; Chen, Q.; et al. UCH-L1 expression of podocytes in diseased glomeruli and in vitro. J. Pathol. 2009, 217, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003, 63, 1164–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Hir, M.; Besse-Eschmann, V. A novel mechanism of nephron loss in a murine model of crescentic glomerulonephritis. Kidney Int. 2003, 63, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Tang, Z.; Xiang, H.; Li, X.; Chen, H.; Zhang, H.; Hu, W.; Zeng, C.; Liu, Z. Etiology and outcome of crescentic glomerulonephritis from a single center in China: A 10-Year Review. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2016, 67, 376–383. [Google Scholar] [CrossRef]
- Chen, S.; Tang, Z.; Zhang, H.; Hu, W.; Liu, Z. Prediction of renal outcomes in patients with crescentic lupus nephritis. Am. J. Med. Sci. 2015, 349, 298–305. [Google Scholar] [CrossRef]
- Rijnink, E.C.; Teng, Y.K.O.; Wilhelmus, S.; Almekinders, M.; Wolterbeek, R.; Cransberg, K.; Bruijn, J.A.; Bajema, I.M. Clinical and Histopathologic Characteristics Associated with Renal Outcomes in Lupus Nephritis. Clin. J. Am. Soc. Nephrol. CJASN 2017, 12, 734–743. [Google Scholar] [CrossRef]
- Cai, F.; Han, F.; Wang, H.; Han, H.; Le, J.; Lan, L.; Xu, Y.; Chen, J. The Crescentic Implication of Renal Outcomes in Proliferative Lupus Nephritis. J. Rheumatol. 2018, 45, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Hénique, C.; Papista, C.; Guyonnet, L.; Lenoir, O.; Tharaux, P.-L. Update on crescentic glomerulonephritis. Semin. Immunopathol. 2014, 36, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.J.; Soofi, A.; Hartmann, I.; Le Hir, M.; Wiggins, R.; Kriz, W.; Holzman, L.B. Podocytes populate cellular crescents in a murine model of inflammatory glomerulonephritis. J. Am. Soc. Nephrol. JASN 2004, 15, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Thorner, P.S.; Ho, M.; Eremina, V.; Sado, Y.; Quaggin, S. Podocytes contribute to the formation of glomerular crescents. J. Am. Soc. Nephrol. JASN 2008, 19, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, M.; Keller, C.; Eschmann, V.; Hähnel, B.; Hosser, H.; Kriz, W. Podocyte bridges between the tuft and Bowman’s capsule: An early event in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. JASN 2001, 12, 2060–2071. [Google Scholar] [PubMed]
- Bariéty, J.; Bruneval, P.; Meyrier, A.; Mandet, C.; Hill, G.; Jacquot, C. Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int. 2005, 68, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Masaki, T.; Hirai, T.; Doi, S.; Kuratsune, M.; Arihiro, K.; Kohno, N.; Yorioka, N. Activation of signal transducer and activator of transcription 3 correlates with cell proliferation and renal injury in human glomerulonephritis. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2008, 23, 3418–3426. [Google Scholar] [CrossRef]
- Dai, Y.; Gu, L.; Yuan, W.; Yu, Q.; Ni, Z.; Ross, M.J.; Kaufman, L.; Xiong, H.; Salant, D.J.; He, J.C.; et al. Podocyte-specific deletion of signal transducer and activator of transcription 3 attenuates nephrotoxic serum-induced glomerulonephritis. Kidney Int. 2013, 84, 950–961. [Google Scholar] [CrossRef]
- Bollée, G.; Flamant, M.; Schordan, S.; Fligny, C.; Rumpel, E.; Milon, M.; Schordan, E.; Sabaa, N.; Vandermeersch, S.; Galaup, A.; et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med. 2011, 17, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Henique, C.; Bollée, G.; Loyer, X.; Grahammer, F.; Dhaun, N.; Camus, M.; Vernerey, J.; Guyonnet, L.; Gaillard, F.; Lazareth, H.; et al. Genetic and pharmacological inhibition of microRNA-92a maintains podocyte cell cycle quiescence and limits crescentic glomerulonephritis. Nat. Commun. 2017, 8, 1829. [Google Scholar] [CrossRef]
- Shankland, S.J.; Smeets, B.; Pippin, J.W.; Moeller, M.J. The emergence of the glomerular parietal epithelial cell. Nat. Rev. Nephrol. 2014, 10, 158–173. [Google Scholar] [CrossRef]
- Smeets, B.; Uhlig, S.; Fuss, A.; Mooren, F.; Wetzels, J.F.M.; Floege, J.; Moeller, M.J. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J. Am. Soc. Nephrol. JASN 2009, 20, 2604–2615. [Google Scholar] [CrossRef] [PubMed]
- Roeder, S.S.; Barnes, T.J.; Lee, J.S.; Kato, I.; Eng, D.G.; Kaverina, N.V.; Sunseri, M.W.; Daniel, C.; Amann, K.; Pippin, J.W.; et al. Activated ERK1/2 increases CD44 in glomerular parietal epithelial cells leading to matrix expansion. Kidney Int. 2017, 91, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Eymael, J.; Sharma, S.; Loeven, M.A.; Wetzels, J.F.; Mooren, F.; Florquin, S.; Deegens, J.K.; Willemsen, B.K.; Sharma, V.; van Kuppevelt, T.H.; et al. CD44 is required for the pathogenesis of experimental crescentic glomerulonephritis and collapsing focal segmental glomerulosclerosis. Kidney Int. 2018, 93, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.J.; Smeets, B. Novel target in the treatment of RPGN: The activated parietal cell. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2013, 28, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Iyoda, M.; Shibata, T.; Wada, Y.; Kuno, Y.; Shindo-Hirai, Y.; Matsumoto, K.; Akizawa, T. Long- and short-term treatment with imatinib attenuates the development of chronic kidney disease in experimental anti-glomerular basement membrane nephritis. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2013, 28, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Iyoda, M.; Shibata, T.; Kawaguchi, M.; Yamaoka, T.; Akizawa, T. Preventive and therapeutic effects of imatinib in Wistar-Kyoto rats with anti-glomerular basement membrane glomerulonephritis. Kidney Int. 2009, 75, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Sasaki, S.; Yamazaki, T.; Sato, Y.; Ito, H.; Ariga, T. Prevalence of CD44-Positive Glomerular Parietal Epithelial Cells Reflects Podocyte Injury in Adriamycin Nephropathy. Nephron Exp. Nephrol. 2013, 124, 11–18. [Google Scholar] [CrossRef]
- Estrada, C.C.; Paladugu, P.; Guo, Y.; Pace, J.; Revelo, M.P.; Salant, D.J.; Shankland, S.J.; D’Agati, V.D.; Mehrotra, A.; Cardona, S.; et al. Krüppel-like factor 4 is a negative regulator of STAT3-induced glomerular epithelial cell proliferation. JCI Insight 2018. [Google Scholar] [CrossRef]
- Romoli, S.; Angelotti, M.L.; Antonelli, G.; Kumar Vr, S.; Mulay, S.R.; Desai, J.; Anguiano Gomez, L.; Thomasova, D.; Eulberg, D.; Klussmann, S.; et al. CXCL12 blockade preferentially regenerates lost podocytes in cortical nephrons by targeting an intrinsic podocyte-progenitor feedback mechanism. Kidney Int. 2018, 94, 1111–1126. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Kobayashi, N.; Nakayama, M.; Takashima, Y.; Ohse, T.; Pastan, I.; Pippin, J.W.; Shankland, S.J.; Uesugi, N.; Matsusaka, T.; et al. Aberrant Notch1-dependent effects on glomerular parietal epithelial cells promotes collapsing focal segmental glomerulosclerosis with progressive podocyte loss. Kidney Int. 2013, 83, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Eng, D.G.; Sunseri, M.W.; Kaverina, N.V.; Roeder, S.S.; Pippin, J.W.; Shankland, S.J. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 999–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohse, T.; Vaughan, M.R.; Kopp, J.B.; Krofft, R.D.; Marshall, C.B.; Chang, A.M.; Hudkins, K.L.; Alpers, C.E.; Pippin, J.W.; Shankland, S.J. De novo expression of podocyte proteins in parietal epithelial cells during experimental glomerular disease. Am. J. Physiol. Ren. Physiol. 2010, 298, F702–F711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machida, H.; Ito, S.; Hirose, T.; Takeshita, F.; Oshiro, H.; Nakamura, T.; Mori, M.; Inayama, Y.; Yan, K.; Kobayashi, N.; et al. Expression of Toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2010, 25, 2530–2537. [Google Scholar] [CrossRef] [PubMed]
- Goldwich, A.; Burkard, M.; Ölke, M.; Daniel, C.; Amann, K.; Hugo, C.; Kurts, C.; Steinkasserer, A.; Gessner, A. Podocytes Are Nonhematopoietic Professional Antigen-Presenting Cells. J. Am. Soc. Nephrol. JASN 2013, 24, 906–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Baye, E.; Gallazzini, M.; Delville, M.; Legendre, C.; Terzi, F.; Canaud, G. The costimulatory receptor B7-1 is not induced in injured podocytes. Kidney Int. 2016, 90, 1037–1044. [Google Scholar] [CrossRef]
- Novelli, R.; Gagliardini, E.; Ruggiero, B.; Benigni, A.; Remuzzi, G. Another Piece of the Puzzle of Podocyte B7-1 Expression: Lupus Nephritis. Nephron 2016, 133, 129–138. [Google Scholar] [CrossRef]
- Furie, R.; Nicholls, K.; Cheng, T.-T.; Houssiau, F.; Burgos-Vargas, R.; Chen, S.-L.; Hillson, J.L.; Meadows-Shropshire, S.; Kinaszczuk, M.; Merrill, J.T. Efficacy and safety of abatacept in lupus nephritis: A twelve-month, randomized, double-blind study. Arthritis Rheumatol. Hoboken NJ 2014, 66, 379–389. [Google Scholar] [CrossRef]
- ACCESS Trial Group. Treatment of lupus nephritis with abatacept: The Abatacept and Cyclophosphamide Combination Efficacy and Safety Study. Arthritis Rheumatol. Hoboken NJ 2014, 66, 3096–3104. [Google Scholar] [CrossRef]
- Ichinose, K.; Ushigusa, T.; Nishino, A.; Nakashima, Y.; Suzuki, T.; Horai, Y.; Koga, T.; Kawashiri, S.; Iwamoto, N.; Tamai, M.; et al. Lupus Nephritis IgG Induction of Calcium/Calmodulin-Dependent Protein Kinase IV Expression in Podocytes and Alteration of Their Function. Arthritis Rheumatol. Hoboken NJ 2016, 68, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.D.; Beresford, M.W. Podocytes contribute, and respond, to the inflammatory environment in lupus nephritis. Am. J. Physiol. Ren. Physiol. 2018, 315, F1683–F1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Guo, C.; Wang, S.; Huang, Y.; Jin, O.; Hu, H.; Chen, J.; Xu, B.; Zhou, M.; Zhao, J.; et al. Podocyte Activation of NLRP3 Inflammasomes Contributes to the Development of Proteinuria in Lupus Nephritis. Arthritis Rheumatol. 2017, 69, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Fu, R.; Zhou, M.; Wang, S.; Huang, Y.; Hu, H.; Zhao, J.; Gaskin, F.; Yang, N.; Fu, S.M. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J. Autoimmun. 2019, 103, 102286. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. JASN 2011, 22, 2129–2137. [Google Scholar] [CrossRef]
- Ashley-Koch, A.E.; Okocha, E.C.; Garrett, M.E.; Soldano, K.; De Castro, L.M.; Jonassaint, J.C.; Orringer, E.P.; Eckman, J.R.; Telen, M.J. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br. J. Haematol. 2011, 155, 386–394. [Google Scholar] [CrossRef]
- Larsen, C.P.; Beggs, M.L.; Saeed, M.; Walker, P.D. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J. Am. Soc. Nephrol. JASN 2013, 24, 722–725. [Google Scholar] [CrossRef]
- Freedman, B.I.; Langefeld, C.D.; Andringa, K.K.; Croker, J.A.; Williams, A.H.; Garner, N.E.; Birmingham, D.J.; Hebert, L.A.; Hicks, P.J.; Segal, M.S.; et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. Hoboken NJ 2014, 66, 390–396. [Google Scholar] [CrossRef]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.D.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; Hu, C.-A.A.; et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, P.; Susztak, K. APOL1: The Balance Imposed by Infection, Selection, and Kidney Disease. Trends Mol. Med. 2018, 24, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Thunø, M.; Macho, B.; Eugen-Olsen, J. suPAR: The Molecular Crystal Ball. Dis. Markers 2009, 27, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Koh, K.H.; Grams, M.E.; Wei, C.; Ko, Y.-A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Giglio, S.; Angelotti, M.L.; Provenzano, A.; Becherucci, F.; Mazzinghi, B.; Müller, S.; Amann, K.; Weidenbusch, M.; Romoli, S.; et al. Next generation sequencing and functional analysis of patient urine renal progenitor-derived podocytes to unravel the diagnosis underlying refractory lupus nephritis. Nephrol. Dial. Transplant. Off. J. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2016, 31, 1541–1545. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. CJASN 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, P.L.; Nolph, K.D.; Jacobs, R.; Luger, A.M.; Walker, S.E. Idiopathic focal segmental glomerulosclerosis in a patient with systemic lupus erythematosus: An unusual combination. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1994, 23, 582–586. [Google Scholar] [CrossRef]
- Papo, T.; Faucher, C.; Huong, D.L.; Beaufils, H.; Piette, J.C.; Godeau, P. Idiopathic focal segmental glomerulosclerosis in a patient with systemic lupus erythematosus: An unusual combination. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1994, 24, 880–881. [Google Scholar] [CrossRef]
- Nishihara, G.; Nakamoto, M.; Yasunaga, C.; Takeda, K.; Matsuo, K.; Urabe, M.; Goya, T.; Sakemi, T. Systemic lupus erythematosus in a patient with remitting minimal change nephrotic syndrome. Clin. Nephrol. 1997, 48, 327–330. [Google Scholar]
- Hertig, A.; Droz, D.; Lesavre, P.; Grünfeld, J.-P.; Rieu, P. SLE and idiopathic nephrotic syndrome: Coincidence or not? Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2002, 40, 1179–1184. [Google Scholar] [CrossRef]
- Dube, G.K.; Markowitz, G.S.; Radhakrishnan, J.; Appel, G.B.; D’Agati, V.D. Minimal change disease in systemic lupus erythematosus. Clin. Nephrol. 2002, 57, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Kraft, S.W.; Schwartz, M.M.; Korbet, S.M.; Lewis, E.J. Glomerular podocytopathy in patients with systemic lupus erythematosus. J. Am. Soc. Nephrol. JASN 2005, 16, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chen, Y.; Wang, S.; Chen, H.; Liu, Z.; Zeng, C.; Zhang, H.; Liu, Z. Clinical-Morphological Features and Outcomes of Lupus Podocytopathy. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Chen, Y.H.; Chen, D.Q.; Liu, Z.Z.; Xu, F.; Zeng, C.H.; Hu, W.X. Mesangial proliferative lupus nephritis with podocytopathy: A special entity of lupus nephritis. Lupus 2018, 27, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Ramée, M.P.; Autuly, V.; Laruelle, E.; Charasse, C.; Cam, G.; Ang, K.S. Epidemiology of primary glomerular diseases in a French region. Variations according to period and age. Kidney Int. 1994, 46, 1192–1198. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Class I | Minimal mesangial lupus nephritis | Light microscopy: normal glomeruli Immunofluorescence: possible mesangial immune deposits |
| Class II | Mesangial proliferative lupus nephritis | Light microscopy: mesangial hypercellularity and/or mesangial matrix immunofluorescence: mesangial immune deposits/possibly a few isolated subepithelial or subendothelial deposits |
| Lupus podocytopathy * | Light microscopy: normal glomeruli/isolated mesangial hypercellularity/focal and segmental glomerulosclerosis (FSGS) Immunofluorescence: no subendothelial or subepithelial deposits, but possible mesangial deposits Electron microscopy: extensive foot process effacement (FPE) | |
| Class III | Focal lupus nephritis | Light microscopy: endo- and/or extracapillary glomerulonephritis involving <50% of all glomeruli Immunofluorescence: subendothelial immune deposits |
| Class IV | Diffuse lupus nephritis | Light microscopy: endo- and/or extracapillary glomerulonephritis involving >50% of all glomeruli Immunofluorescence: subendothelial immune deposits |
| Class V | Membranous lupus nephritis | Light microscopy: Morphological aspects of membranous nephropathy. Immunofluorescence: subepithelial immune deposits Possible combination with class III or IV |
| Class VI | Advanced sclerotic lupus nephritis | ≥90% of glomeruli sclerotic |
| Author, Year N. pat. | Inclusion Criteria | Clinical Presentation | Histology | Prognosis | |||
|---|---|---|---|---|---|---|---|
| Light Microscopy | Ig Deposits | FPE | Response | Relapse and Hist. Trans. | |||
| Hertig, 2002 [100] n = 11 |
| Female 27 years NS: 100%Hu: 30%AKI: 55% Time onset LES/NS: 5 months | Ms proliferation: 0% MCD: 30% FSGS: 70% | Ms deposit: 30% | - | CR: 70% | Relapse: 30% Hist. Trans.: - |
| Kraft, 2005 [102] n = 18 |
| Female 30 years NS: 45%Hu: 60%AKI: 20% Time onset LES/NS: 1 month (in NS range) | Ms proliferation: 75% MCD: 15% FSGS: 45% | Ms deposit: 75% | FPE: >80% in 90% of NS | CR/PR: 75% | Relapse: - |
| Hu, 2016 [103] n = 50 |
| Female 30 years NS: 100%Hu: 20–30%AKI: Class I/II appearance: 20%; FSGS 70% Time onset LES/NS: 5 months | Ms proliferation: 74% MCD: 26% FSGS: 18% | Ms deposit: 90% | FPE: >70% in 90% of patients | CR: 75%PR: 20% | Relapse: 50% Hist. Trans.: 40% |
| Wang, 2017 [104] n = 31 |
| Female 30 years NS: 100% Hu: 20%AKI: 30% Time onset LES/NS: 6 months | Ms proliferation: 100% | Ms deposit: 100% | FPE: >70% in 75% of patients | CR: 85%PR: 10% | Relapse: 50% Hist. Trans: 60% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakhi, H.; Moktefi, A.; Bouachi, K.; Audard, V.; Hénique, C.; Remy, P.; Ollero, M.; El Karoui, K. Podocyte Injury in Lupus Nephritis. J. Clin. Med. 2019, 8, 1340. https://doi.org/10.3390/jcm8091340
Sakhi H, Moktefi A, Bouachi K, Audard V, Hénique C, Remy P, Ollero M, El Karoui K. Podocyte Injury in Lupus Nephritis. Journal of Clinical Medicine. 2019; 8(9):1340. https://doi.org/10.3390/jcm8091340
Chicago/Turabian StyleSakhi, Hamza, Anissa Moktefi, Khedidja Bouachi, Vincent Audard, Carole Hénique, Philippe Remy, Mario Ollero, and Khalil El Karoui. 2019. "Podocyte Injury in Lupus Nephritis" Journal of Clinical Medicine 8, no. 9: 1340. https://doi.org/10.3390/jcm8091340
APA StyleSakhi, H., Moktefi, A., Bouachi, K., Audard, V., Hénique, C., Remy, P., Ollero, M., & El Karoui, K. (2019). Podocyte Injury in Lupus Nephritis. Journal of Clinical Medicine, 8(9), 1340. https://doi.org/10.3390/jcm8091340