Early Nuclear Events after Herpesviral Infection


{kind=link}
{kind=link}
Abstract
1. Introduction
2. DNA Sensor Proteins
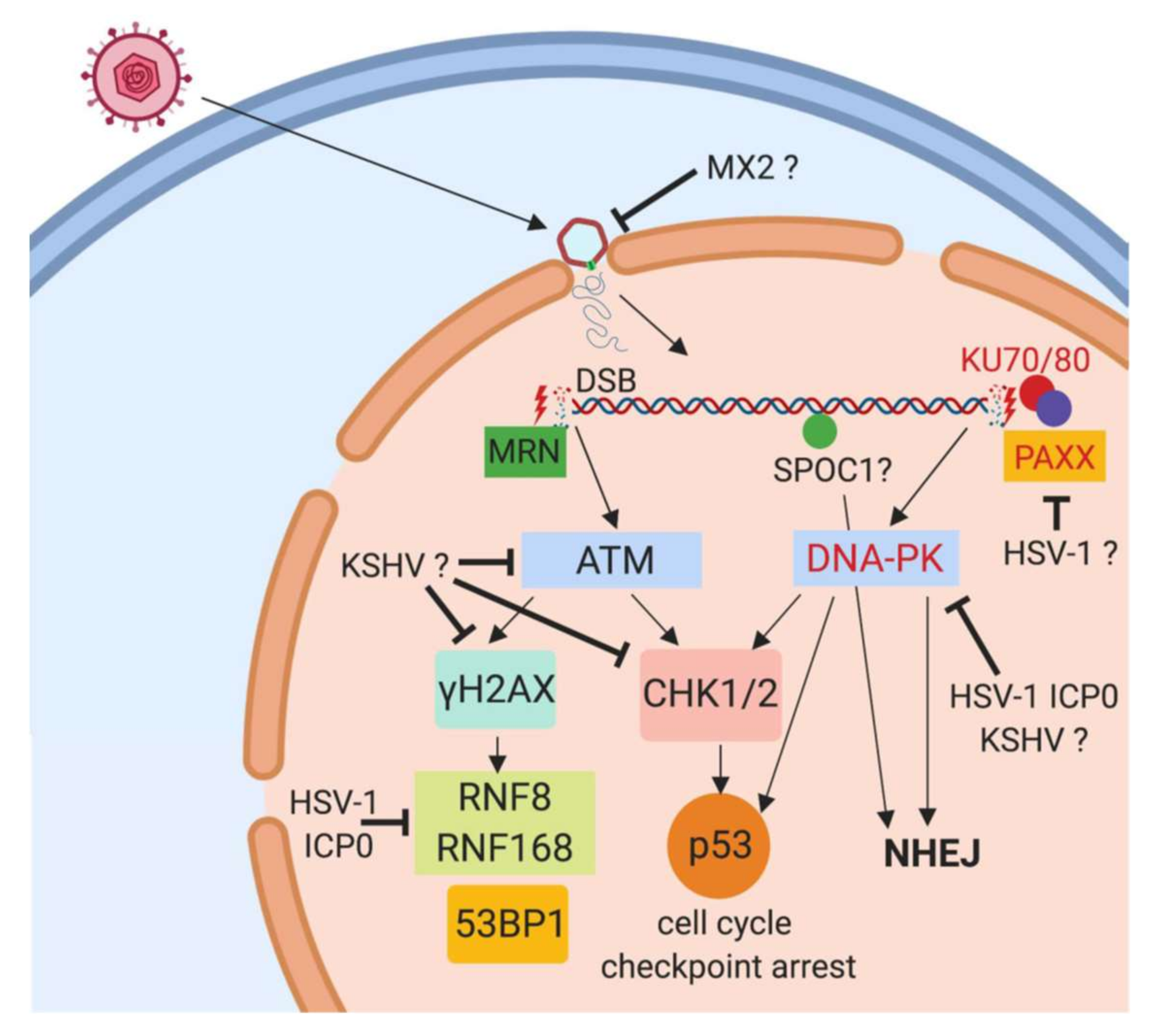
3. DNA Damage Response Proteins
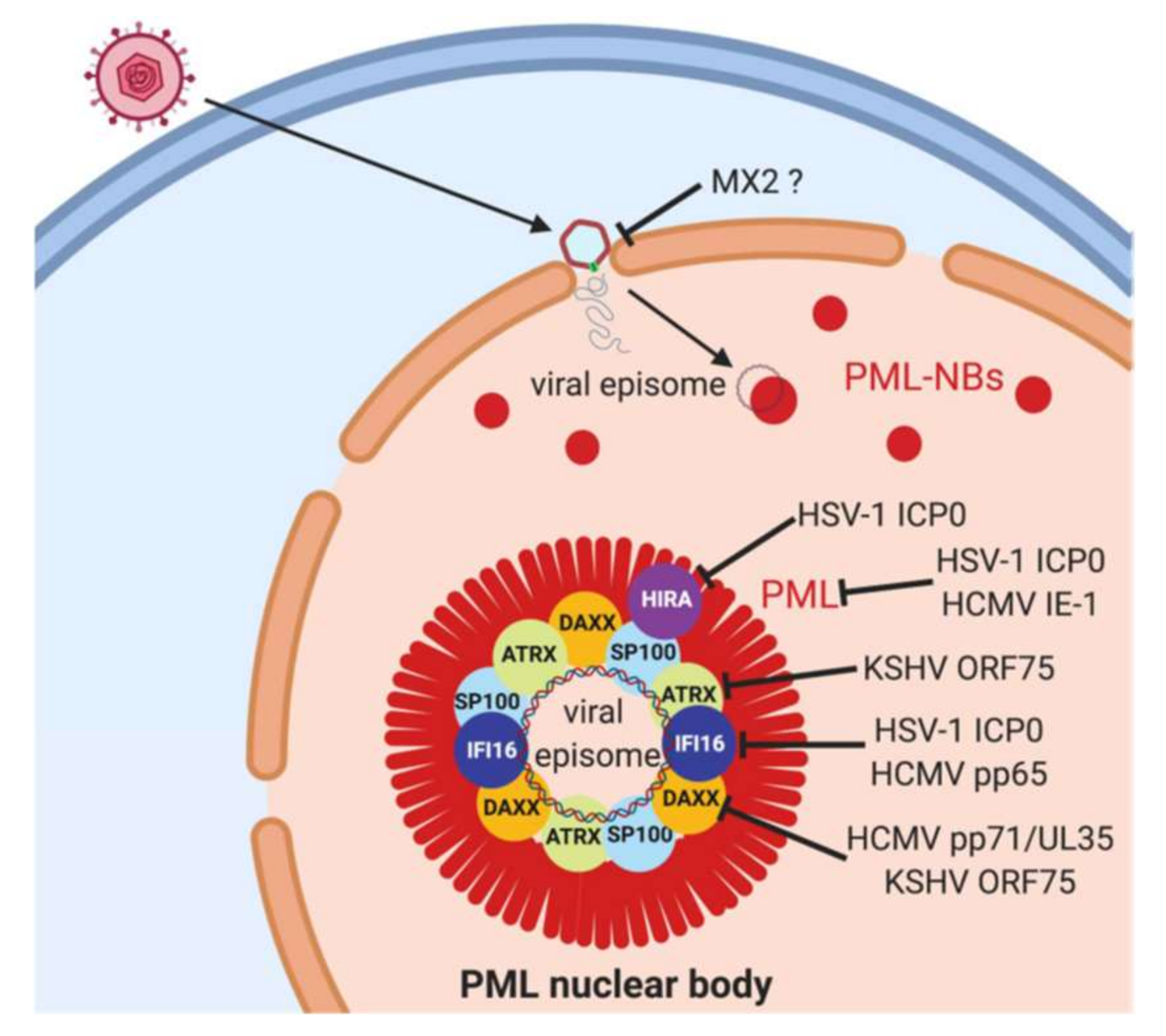
4. PML Nuclear Bodies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pellet, P.E.; Roizman, B. Fields Virology; Wolters Kluwer: Alfon am Rhein, The Netherlands, 2013. [Google Scholar]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef] [PubMed]
- Emery, V.C.; Clark, D.A. HHV-6A, 6B, and 7: Persistence in the population, epidemiology and transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Hjalgrim, H.; Friborg, J.; Melbye, M. The epidemiology of EBV and its association with malignant disease. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Liu, Y.T.; Jih, J.; Dai, X.; Bi, G.Q.; Zhou, Z.H. Cryo-EM structures of herpes simplex virus type 1 portal vertex and packaged genome. Nature 2019, 570, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 2015, 479, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Drayman, N.; Butin-Israeli, V.; Oppenheim, A. Virus strategies for passing the nuclear envelope barrier. Nucleus 2012, 3, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef]
- Komatsu, T.; Nagata, K.; Wodrich, H. The Role of Nuclear Antiviral Factors against Invading DNA Viruses: The Immediate Fate of Incoming Viral Genomes. Viruses 2016, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Iqbal, J.; Kumar, B.; Roy, A.; Chikoti, L.; Singh, V.V.; Chandran, B. Herpesvirus Genome Recognition Induced Acetylation of Nuclear IFI16 Is Essential for Its Cytoplasmic Translocation, Inflammasome and IFN-beta Responses. PLoS Pathog. 2015, 11, e1005019. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Dynamic Response of IFI16 and Promyelocytic Leukemia Nuclear Body Components to Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Lum, K.K.; Howard, T.R.; Pan, C.; Cristea, I.M. Charge-Mediated Pyrin Oligomerization Nucleates Antiviral IFI16 Sensing of Herpesvirus DNA. MBio 2019. [Google Scholar] [CrossRef] [PubMed]
- Merkl, P.E.; Knipe, D.M. Role for a Filamentous Nuclear Assembly of IFI16, DNA, and Host Factors in Restriction of Herpesviral Infection. MBio 2019. [Google Scholar] [CrossRef]
- Gariano, G.R.; Dell’Oste, V.; Bronzini, M.; Gatti, D.; Luganini, A.; De Andrea, M.; Gribaudo, G.; Gariglio, M.; Landolfo, S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012, 8, e1002498. [Google Scholar] [CrossRef]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe. 2013, 14, 591–599. [Google Scholar] [CrossRef]
- Cuchet-Lourenco, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell Proteomics. 2015, 14, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-beta Responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef]
- Iqbal, J.; Ansari, M.A.; Kumar, B.; Dutta, D.; Roy, A.; Chikoti, L.; Pisano, G.; Dutta, S.; Vahedi, S.; Veettil, M.V.; et al. Histone H2B-IFI16 Recognition of Nuclear Herpesviral Genome Induces Cytoplasmic Interferon-beta Responses. PLoS Pathog. 2016, 12, e1005967. [Google Scholar] [CrossRef]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef]
- Alandijany, T.; Roberts, A.P.E.; Conn, K.L.; Loney, C.; McFarlane, S.; Orr, A.; Boutell, C. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLoS Pathog. 2018, 14, e1006769. [Google Scholar] [CrossRef]
- Komatsu, T.; Nagata, K.; Wodrich, H. An Adenovirus DNA Replication Factor, but Not Incoming Genome Complexes, Targets PML Nuclear Bodies. J. Virol. 2016, 90, 1657–1667. [Google Scholar] [CrossRef]
- Komatsu, T.; Will, H.; Nagata, K.; Wodrich, H. Imaging analysis of nuclear antiviral factors through direct detection of incoming adenovirus genome complexes. Biochem. Biophys. Res. Commun. 2016, 473, 200–205. [Google Scholar] [CrossRef]
- Cabral, J.M.; Oh, H.S.; Knipe, D.M. ATRX promotes maintenance of herpes simplex virus heterochromatin during chromatin stress. Elife 2018. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Knipe, D.M. Cellular sensing of viral DNA and viral evasion mechanisms. Annu. Rev. Microbiol. 2014, 68, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.A.; O’Shea, C.C. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Rohleder, K.J.; Hanakahi, L.A.; Ketner, G. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J. Virol. 2007, 81, 7034–7040. [Google Scholar] [CrossRef]
- Schrank, B.; Gautier, J. Assembling nuclear domains: Lessons from DNA repair. J. Cell Biol. 2019. [Google Scholar] [CrossRef]
- Smith, S.; Reuven, N.; Mohni, K.N.; Schumacher, A.J.; Weller, S.K. Structure of the herpes simplex virus 1 genome: Manipulation of nicks and gaps can abrogate infectivity and alter the cellular DNA damage response. J. Virol. 2014, 88, 10146–10156. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef]
- Flamand, L. Chromosomal Integration by Human Herpesviruses 6A and 6B. Adv. Exp. Med. Biol. 2018, 1045, 209–226. [Google Scholar] [PubMed]
- Nacheva, E.P.; Ward, K.N.; Brazma, D.; Virgili, A.; Howard, J.; Leong, H.N.; Clark, D.A. Human herpesvirus 6 integrates within telomeric regions as evidenced by five different chromosomal sites. J. Med. Virol. 2008, 80, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Dee, A.R.; Smith, S.; Schumacher, A.J.; Weller, S.K. Efficient herpes simplex virus 1 replication requires cellular ATR pathway proteins. J. Virol. 2013, 87, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Livingston, C.M.; Cortez, D.; Weller, S.K. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J. Virol. 2010, 84, 12152–12164. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Bloom, D.C.; Fisher, C. The ATM and Rad3-Related (ATR) Protein Kinase Pathway Is Activated by Herpes Simplex Virus 1 and Required for Efficient Viral Replication. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Albright, B.S.; Kosinski, A.; Szczepaniak, R.; Cook, E.A.; Stow, N.D.; Conway, J.F.; Weller, S.K. The putative herpes simplex virus 1 chaperone protein UL32 modulates disulfide bond formation during infection. J. Virol. 2015, 89, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Smith, S.; Dee, A.R.; Schumacher, A.J.; Weller, S.K. Herpes simplex virus type 1 single strand DNA binding protein and helicase/primase complex disable cellular ATR signaling. PLoS Pathog. 2013, 9, e1003652. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Weller, S.K. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Schwartz, R.A.; Weitzman, M.D. Using or abusing: Viruses and the cellular DNA damage response. Trends. Microbiol. 2007, 15, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; Dremel, S.E.; DeLuca, N.A. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017, 13, e1006166. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Horniblow, R.D.; Forrest, C.; Stewart, G.S.; Grand, R.J. Localization of Double-Strand Break Repair Proteins to Viral Replication Compartments following Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Skalka, G.L.; Stewart, G.S.; Hislop, A.D.; Blackbourn, D.J.; Grand, R.J. Activation of DNA Damage Response Pathways during Lytic Replication of KSHV. Viruses 2015, 7, 2908–2927. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, J.M.; Zavala, A.G.; Brown, C.J.; Mori, T.; Fortunato, E.A. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 2012, 8, e1003038. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Rosenke, K.; Czornak, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.P.; Frame, F.M.; Rogoff, H.A.; Pickering, M.T.; Yurochko, A.D.; Kowalik, T.F. Human cytomegalovirus IE1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J. Virol. 2005, 79, 11467–11475. [Google Scholar] [CrossRef] [PubMed]
- Xiaofei, E.; Pickering, M.T.; Debatis, M.; Castillo, J.; Lagadinos, A.; Wang, S.; Lu, S.; Kowalik, T.F. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 2011, 7, e1001342. [Google Scholar] [CrossRef]
- Xiaofei, E.; Savidis, G.; Chin, C.R.; Wang, S.; Lu, S.; Brass, A.L.; Kowalik, T.F. A novel DDB2-ATM feedback loop regulates human cytomegalovirus replication. J. Virol. 2014, 88, 2279–2290. [Google Scholar]
- Xiaofei, E.; Kowalik, T.F. The DNA damage response induced by infection with human cytomegalovirus and other viruses. Viruses 2014, 6, 2155–2185. [Google Scholar]
- Spector, D.H. Human cytomegalovirus riding the cell cycle. Med. Microbiol. Immunol. 2015, 204, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Trigg, B.J.; Lauer, K.B.; Fernandes Dos Santos, P.; Coleman, H.; Balmus, G.; Mansur, D.S.; Ferguson, B.J. The Non-Homologous End Joining Protein PAXX Acts to Restrict HSV-1 Infection. Viruses 2017, 9, 342. [Google Scholar] [CrossRef] [PubMed]
- Reichel, A.; Stilp, A.C.; Scherer, M.; Reuter, N.; Lukassen, S.; Kasmapour, B.; Schreiner, S.; Cicin-Sain, L.; Winterpacht, A.; Stamminger, T. Chromatin-Remodeling Factor SPOC1 Acts as a Cellular Restriction Factor against Human Cytomegalovirus by Repressing the Major Immediate Early Promoter. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Kinkley, S.; Burck, C.; Mund, A.; Wimmer, P.; Schubert, T.; Groitl, P.; Will, H.; Dobner, T. SPOC1-mediated antiviral host cell response is antagonized early in human adenovirus type 5 infection. PLoS Pathog. 2013, 9, e1003775. [Google Scholar] [CrossRef] [PubMed]
- Mund, A.; Schubert, T.; Staege, H.; Kinkley, S.; Reumann, K.; Kriegs, M.; Fritsch, L.; Battisti, V.; Ait-Si-Ali, S.; Hoffbeck, A.S.; et al. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic. Acids. Res. 2012, 40, 11363–11379. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.A.; DeLuca, N.A. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. USA 2003, 100, 7871–7876. [Google Scholar] [CrossRef]
- Strang, B.L.; Stow, N.D. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 2005, 79, 12487–12494. [Google Scholar] [CrossRef]
- Skaliter, R.; Lehman, I.R. Rolling circle DNA replication in vitro by a complex of herpes simplex virus type 1-encoded enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 10665–10669. [Google Scholar] [CrossRef]
- Skaliter, R.; Makhov, A.M.; Griffith, J.D.; Lehman, I.R. Rolling circle DNA replication by extracts of herpes simplex virus type 1-infected human cells. J. Virol. 1996, 70, 1132–1136. [Google Scholar]
- Zhang, X.; Efstathiou, S.; Simmons, A. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: Implications for viral DNA amplification strategies. Virology 1994, 202, 530–539. [Google Scholar] [CrossRef]
- Severini, A.; Morgan, A.R.; Tovell, D.R.; Tyrrell, D.L. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology 1994, 200, 428–435. [Google Scholar] [CrossRef]
- Severini, A.; Scraba, D.G.; Tyrrell, D.L. Branched structures in the intracellular DNA of herpes simplex virus type 1. J. Virol. 1996, 70, 3169–3175. [Google Scholar] [PubMed]
- Wilkinson, D.E.; Weller, S.K. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 2003, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; DeLuca, N.A. Temporal Viral Genome-Protein Interactions Define Distinct Stages of Productive Herpesviral Infection. MBio 2018. [Google Scholar] [CrossRef] [PubMed]
- Ligasova, A.; Liboska, R.; Friedecky, D.; Micova, K.; Adam, T.; Ozdian, T.; Rosenberg, I.; Koberna, K. Dr Jekyll and Mr Hyde: A strange case of 5-ethynyl-2’-deoxyuridine and 5-ethynyl-2’-deoxycytidine. Open Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeier, F.; Maya-Mendoza, A.; Jackson, D.A. EdU induces DNA damage response and cell death in mESC in culture. Chromosome. Res. 2013, 21, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.; Lees-Miller, S.P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 1999, 73, 650–657. [Google Scholar] [PubMed]
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 1996, 70, 7471–7477. [Google Scholar] [PubMed]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Stamminger, T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta. 2008, 1783, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Full, F.; Hahn, A.S.; Grosskopf, A.K.; Ensser, A. Gammaherpesviral Tegument Proteins, PML-Nuclear Bodies and the Ubiquitin-Proteasome System. Viruses 2017, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Schilling, E.M.; Stamminger, T. The Human CMV IE1 Protein: An Offender of PML Nuclear Bodies. Adv. Anat. Embryol. Cell Biol. 2017, 223, 77–94. [Google Scholar] [PubMed]
- Schreiner, S.; Burck, C.; Glass, M.; Groitl, P.; Wimmer, P.; Kinkley, S.; Mund, A.; Everett, R.D.; Dobner, T. Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic. Acids. Res. 2013, 41, 3532–3550. [Google Scholar] [CrossRef]
- Albright, E.R.; Kalejta, R.F. Canonical and Variant Forms of Histone H3 Are Deposited onto the Human Cytomegalovirus Genome during Lytic and Latent Infections. J. Virol. 2016, 90, 10309–10320. [Google Scholar] [CrossRef]
- Tsai, K.; Chan, L.; Gibeault, R.; Conn, K.; Dheekollu, J.; Domsic, J.; Marmorstein, R.; Schang, L.M.; Lieberman, P.M. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J. Virol. 2014, 88, 14350–14363. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.; Orr, A.; Roberts, A.P.E.; Conn, K.L.; Iliev, V.; Loney, C.; da Silva Filipe, A.; Smollett, K.; Gu, Q.; Robertson, N.; et al. The histone chaperone HIRA promotes the induction of host innate immune defences in response to HSV-1 infection. PLoS Pathog. 2019, 15, e1007667. [Google Scholar] [CrossRef] [PubMed]
- Rai, T.S.; Glass, M.; Cole, J.J.; Rather, M.I.; Marsden, M.; Neilson, M.; Brock, C.; Humphreys, I.R.; Everett, R.D.; Adams, P.D. Histone chaperone HIRA deposits histone H3.3 onto foreign viral DNA and contributes to anti-viral intrinsic immunity. Nucleic. Acids. Res. 2017, 45, 11673–11683. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Raja, P.; Knipe, D.M. Herpesviral ICP0 Protein Promotes Two Waves of Heterochromatin Removal on an Early Viral Promoter during Lytic Infection. MBio 2016, 7, e02007–e02015. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Stenberg, R.M.; Maul, G.G. Human cytomegalovirus immediate early interaction with host nuclear structures: Definition of an immediate transcript environment. J. Cell Biol. 1997, 138, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, A.; Paulus, C.; Nevels, M. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 2008, 82, 11167–11180. [Google Scholar] [CrossRef] [PubMed]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G.; Ishov, A.M.; Everett, R.D. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 1996, 217, 67–75. [Google Scholar] [CrossRef]
- Scherer, M.; Stamminger, T. Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef]
- Herrera, F.J.; Triezenberg, S.J. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef]
- Maul, G.G.; Everett, R.D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 1994, 75, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar] [PubMed]
- Glass, M.; Everett, R.D. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 2013, 87, 2174–2185. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Vladimirova, O.V.; Maul, G.G. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 2002, 76, 7705–7712. [Google Scholar] [CrossRef] [PubMed]
- Korioth, F.; Maul, G.G.; Plachter, B.; Stamminger, T.; Frey, J. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 1996, 229, 155–158. [Google Scholar] [CrossRef]
- Schilling, E.M.; Scherer, M.; Reuter, N.; Schweininger, J.; Muller, Y.A.; Stamminger, T. The Human Cytomegalovirus IE1 Protein Antagonizes PML Nuclear Body-Mediated Intrinsic Immunity via the Inhibition of PML De Novo SUMOylation. J. Virol. 2017, 91, e02049-16. [Google Scholar] [CrossRef]
- Ensser, A.; Pflanz, R.; Fleckenstein, B. Primary structure of the alcelaphine herpesvirus 1 genome. J. Virol. 1997, 71, 6517–6525. [Google Scholar]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Sturzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed]
- Full, F.; Reuter, N.; Zielke, K.; Stamminger, T.; Ensser, A. Herpesvirus saimiri antagonizes nuclear domain 10-instituted intrinsic immunity via an ORF3-mediated selective degradation of cellular protein Sp100. J. Virol. 2012, 86, 3541–3553. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.S.; Grosskopf, A.K.; Jungnickl, D.; Scholz, B.; Ensser, A. Viral FGARAT Homolog ORF75 of Rhesus Monkey Rhadinovirus Effects Proteasomal Degradation of the ND10 Components SP100 and PML. J. Virol. 2016, 90, 8013–8028. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, J.; Song, S.; He, X.; Minassian, A.; Zhou, Y.; Zhang, J.; Brulois, K.; Wang, Y.; Cabo, J.; et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 2015, 58, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, J.; Xu, S.; Feng, P. Emerging Roles of Protein Deamidation in Innate Immune Signaling. J. Virol. 2016, 90, 4262–4268. [Google Scholar] [CrossRef]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W.; et al. A Viral Deamidase Targets the Helicase Domain of RIG-I to Block RNA-Induced Activation. Cell Host Microbe. 2016, 20, 770–784. [Google Scholar] [CrossRef]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, Z.; Tutton, S.; Lieberman, P.M. The Telomeric Response to Viral Infection. Viruses 2017, 9, 218. [Google Scholar] [CrossRef]
- Merkl, P.E.; Orzalli, M.H.; Knipe, D.M. Mechanisms of Host IFI16, PML, and Daxx Protein Restriction of Herpes Simplex Virus 1 Replication. J. Virol. 2018. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Shire, K.; Wong, A.I.; Tatham, M.H.; Anderson, O.F.; Ripsman, D.; Gulstene, S.; Moffat, J.; Hay, R.T.; Frappier, L. Identification of RNF168 as a PML nuclear body regulator. J. Cell Sci. 2016, 129, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Schierling, K.; Stamminger, T.; Mertens, T.; Winkler, M. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J. Virol. 2004, 78, 9512–9523. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Wang, X.; Frappier, L. Nuclear body formation and PML body remodeling by the human cytomegalovirus protein UL35. Virology 2011, 414, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Jagannathan, M.; Paladino, P.; Chan, P.K.; Dellaire, G.; Raught, B.; Frappier, L. Proteomic profiling of the human cytomegalovirus UL35 gene products reveals a role for UL35 in the DNA repair response. J. Virol. 2012, 86, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Bulli, L.; Weigang, S.; Graf, L.; Naumann, S.; Patzina, C.; Wagner, V.; Bauersfeld, L.; Goujon, C.; Hengel, H.; et al. Human MxB Protein Is a Pan-herpesvirus Restriction Factor. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Crameri, M.; Bauer, M.; Caduff, N.; Walker, R.; Steiner, F.; Franzoso, F.D.; Gujer, C.; Boucke, K.; Kucera, T.; Zbinden, A.; et al. MxB is an interferon-induced restriction factor of human herpesviruses. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- Jaguva Vasudevan, A.A.; Bahr, A.; Grothmann, R.; Singer, A.; Haussinger, D.; Zimmermann, A.; Munk, C. MXB inhibits murine cytomegalovirus. Virology 2018, 522, 158–167. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Full, F.; Ensser, A. Early Nuclear Events after Herpesviral Infection. J. Clin. Med. 2019, 8, 1408. https://doi.org/10.3390/jcm8091408
Full F, Ensser A. Early Nuclear Events after Herpesviral Infection. Journal of Clinical Medicine. 2019; 8(9):1408. https://doi.org/10.3390/jcm8091408
Chicago/Turabian StyleFull, Florian, and Armin Ensser. 2019. "Early Nuclear Events after Herpesviral Infection" Journal of Clinical Medicine 8, no. 9: 1408. https://doi.org/10.3390/jcm8091408
APA StyleFull, F., & Ensser, A. (2019). Early Nuclear Events after Herpesviral Infection. Journal of Clinical Medicine, 8(9), 1408. https://doi.org/10.3390/jcm8091408