Hepatobiliary Complications in Children with Sickle Cell Disease: A Retrospective Review of Medical Records from 616 Patients

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
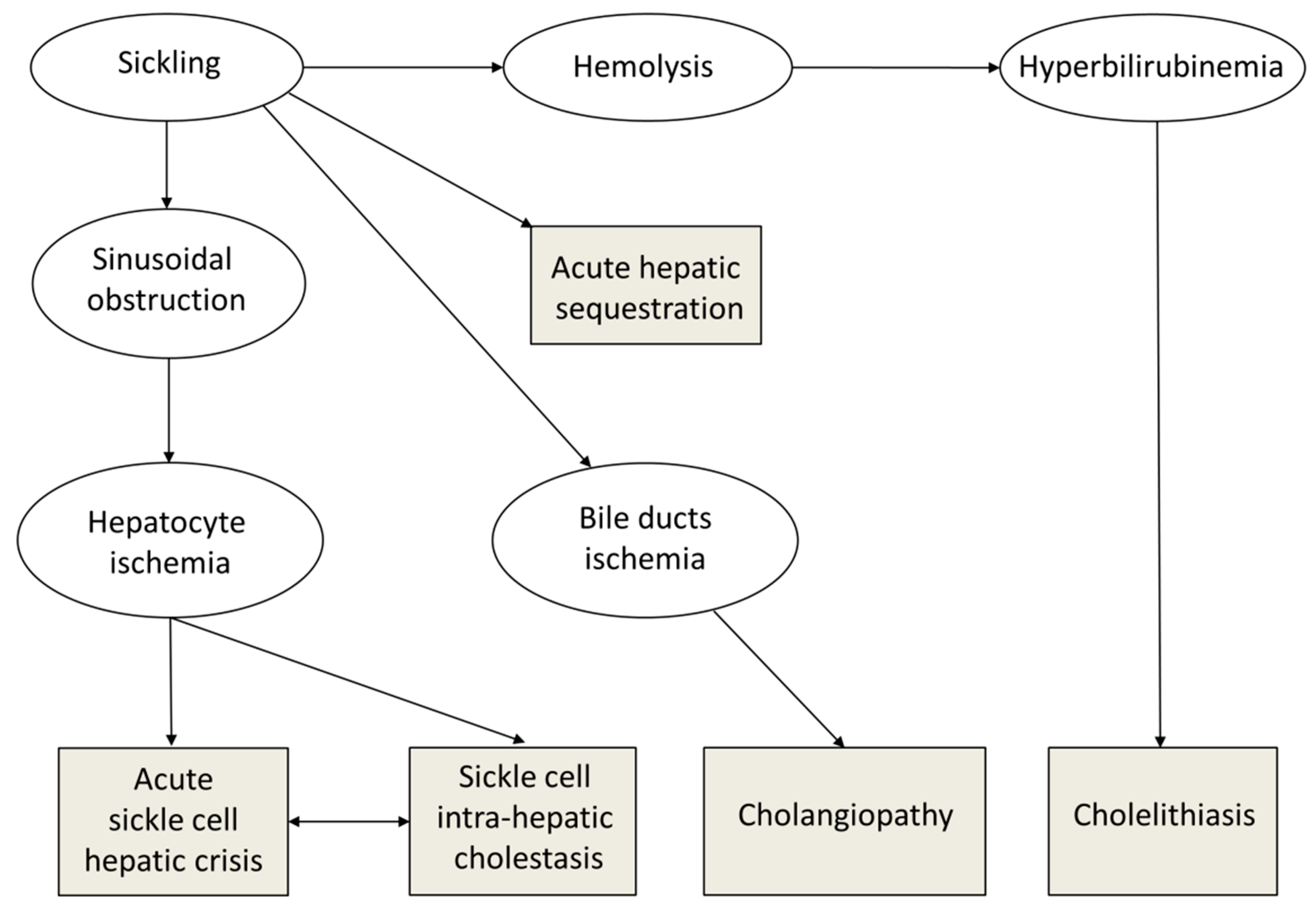
2.2. Classification of Hepatobiliary Complications
- Biliary complications included cholelithiasis, cholecystitis, and cholangitis. Distinction was made between non-migrating lithiasis (some diagnosed after abdominal pain without radiological signs of migration) and migrating lithiasis diagnosed by ultrasonography.
- Cholangiopathy was defined as abnormal bile ducts with stenosis and dilations on ultrasonography and MR cholangiography, and elevated gamma-glutamyltransferase (γ-GT) level. Presence of antineutrophil cytoplasmic antibodies (ANCAs) or associated autoimmune hepatitis was classified as primary sclerosing cholangitis (PSC) [14].
- Auto-immune hepatitis was defined as elevated levels of transaminases (alanine aminotransferase (ALT) level more than twice the upper limit of normal) with hypergammaglobulinemia, positive autoantibodies (antinuclear antibodies (ANAs), anti-smooth muscle antibodies (SMAs), or anti-liver-kidney microsomes antibodies type 1 (LKM-1)), and compatible histopathology (portal inflammation and interface hepatitis).
- Acute sickle cell hepatic crisis was defined by the acute onset of right upper-quadrant abdominal pain, possibly associated with increased hepatomegaly and jaundice, with ALT level more than twice the upper limit of normal or elevated bilirubin level with predominantly conjugated fraction, with no other cause (virus, toxic, gallstone complication). Aspartate aminotransferase (AST) level was not considered for the diagnosis of liver disease because it depends also on hemolysis. We classified “sickle cell intrahepatic cholestasis” as the severe variant of acute hepatic crisis, with coagulopathy, and a possible evolution towards multiple organ failure, as previously published [6].
- Acute hepatic sequestration was defined by the sudden increase in liver size, associated with right upper-quadrant abdominal pain, and an acute decrease in Hb level >2 g/dL. Acute anemia was usually associated with thrombocytopenia and normal or increased reticulocyte count. Because it could be associated with increased transaminase levels, increased conjugated bilirubin level and liver failure, acute hepatic sequestration could also be considered a variant of acute hepatic crisis.
- Transfusion-related liver iron overload was diagnosed by markedly increased ferritin level with MRI-measured liver iron content (LIC) >3 mg iron/g dry weight.
- Hepatotoxicity of chelator drugs was evoked on abnormal liver tests after excluding other causes. Some patients underwent a reintroduction challenge: drug was reintroduced at a lower dose and liver tests were controlled.
- Infectious hepatitis was diagnosed on abnormal liver tests with the identification of a causative infectious agent (mainly viruses).
- Isolated biochemical abnormalities were defined as increased ALT level more than twice the upper limit of normal, elevated conjugated bilirubin level, or elevated γ-GT level, in the absence of any symptom.
2.3. Statistical Analysis
3. Results
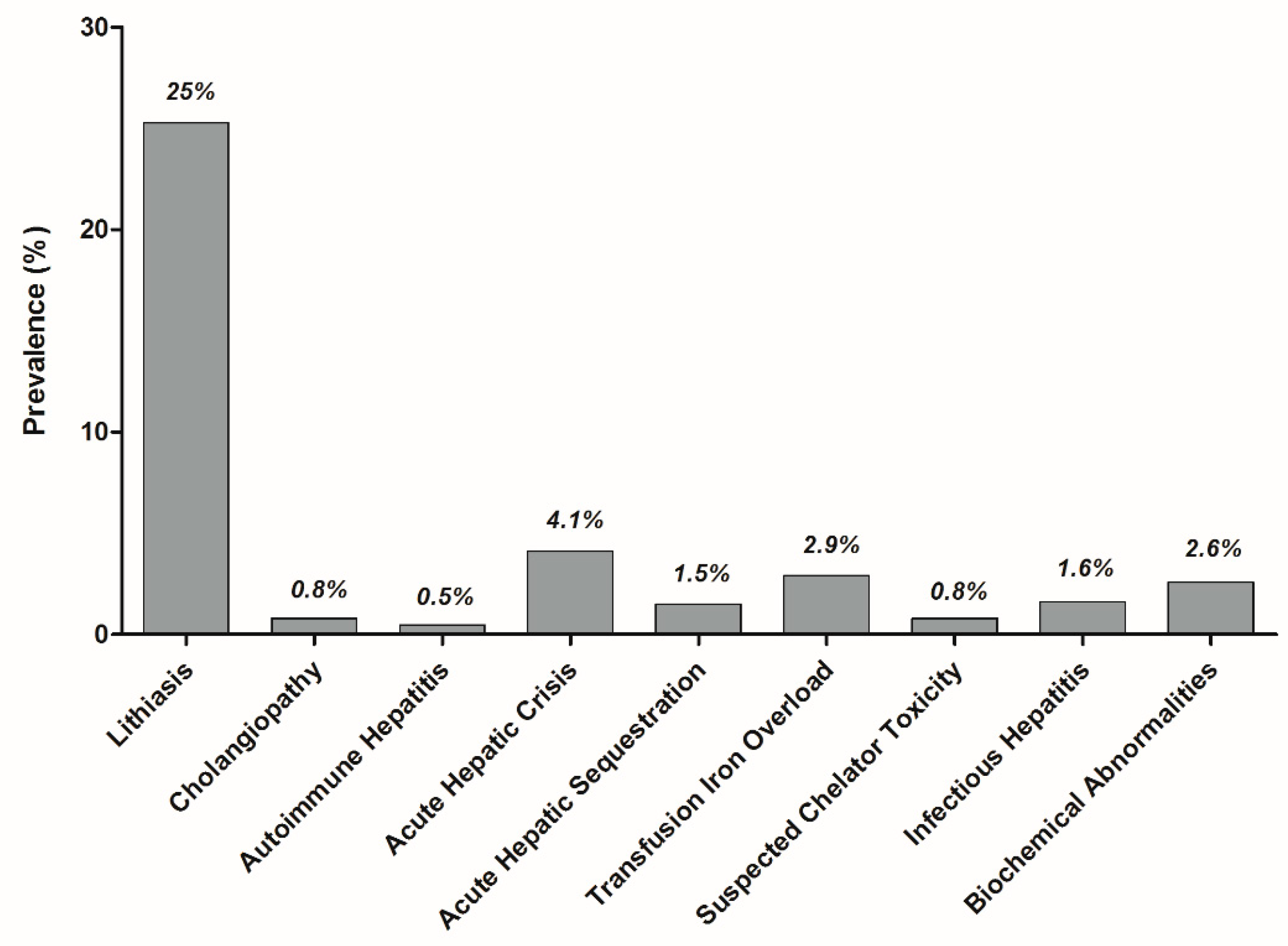
3.1. Cholelithiasis
3.2. Cholangiopathy
3.3. Auto-Immune Hepatitis
3.4. Acute Sickle Cell Hepatic Crisis, Sickle Cell Intrahepatic Cholestasis, and Acute Hepatic Sequestration
3.5. Transfusion Iron Overload
3.6. Hepatotoxicity of Oral Chelators
3.7. Infectious Hepatitis
3.8. Isolated Liver Tests Abnormalities
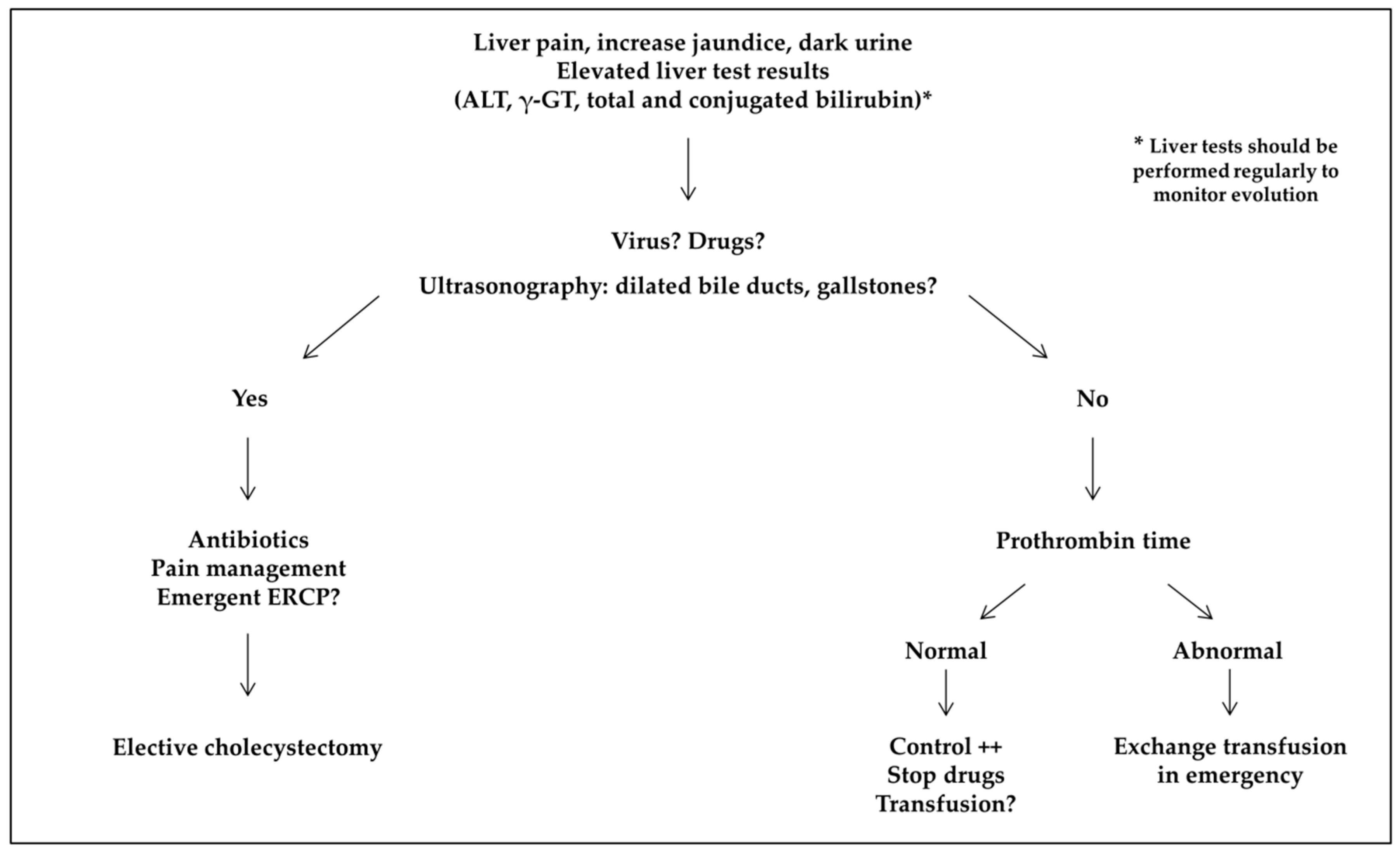
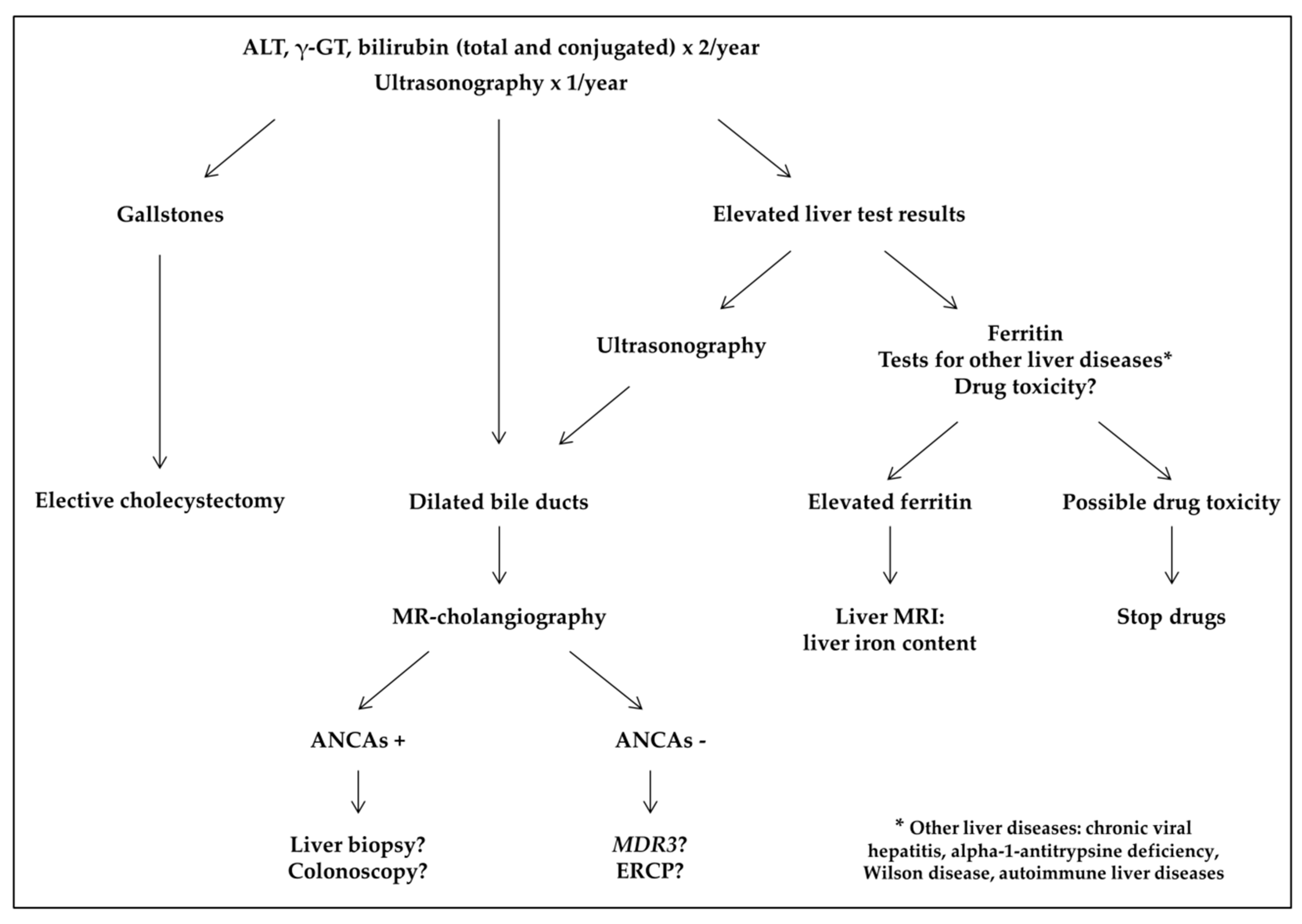
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle cell disease. N. Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Owen, C.; Chopra, S. Sickle cell hepatopathy. Hepatology 2001, 33, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Taborda, C.; Chawla, S. Acute and chronic hepatobiliary manifestations of sickle cell disease: A review. World J. Gastrointest. Pathophysiol. 2017, 8, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Schubert, T.T. Hepatobiliary system in sickle cell disease. Gastroenterology 1986, 90, 2013–2021. [Google Scholar] [CrossRef]
- Ware, R.E.; De Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 309, 311–323. [Google Scholar] [CrossRef]
- Gardner, K.; Suddie, A.; Kane, P.; O’Grady, J.; Heaton, N.; Bomford, A.; Thein, S.L. How we treat sickle hepatopathy and liver transplantation in adults. Blood 2014, 123, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef]
- Ebert, E.C.; Nagar, M.; Hagspiel, K.D. Gastrointestinal and hepatic complications of sickle cell disease. Clin. Gastroenterol. Hepatol. 2010, 8, 483–489. [Google Scholar] [CrossRef]
- Almeida, R.P.; Ferreira, C.D.; Conceicao, J.; Franca, R.; Lyra, I.; Rodrigues Silva, L. Hepatobiliary abnormalities in pediatric patients with sickle cell disease. Acta Gastroenterol. Latinoam. 2009, 39, 112–117. [Google Scholar]
- Lacaille, F.; Lesage, F.; De Montalembert, M. Acute hepatic crisis in children with sickle cell disease. J. Pediatr. Gastroenterol. Nutr. 2004, 39, 200–202. [Google Scholar] [CrossRef]
- Pecker, L.H.; Patel, N.; Creary, N.; Darbari, A.; Riehm Meier, E.; Darbari, D.S.; Fasano, R.M. Diverse manifestations of acute sickle cell hepatopathy in pediatric patients with sickle cell disease: A case series. Pediatr. Blood Cancer 2018, 65, e270060. [Google Scholar] [CrossRef] [PubMed]
- Jitraruch, A.; Fitzpatrick, E.; Deheragoda, M.; Deganello, A.; Mieilli-Vergani, G.; Height, S.; Rees, D.; Hadzic, N.; Samyn, M. Autoimmune liver disease in children with sickle cell disease. J. Pediatr. 2017, 189, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Garcelon, N.; Neuraz, A.; Salomon, R.; Faour, H.; Benoit, V.; Delapalme, A.; Munnich, A.; Burgun, A.; Rancé, B. A clinician friendly data warehouse oriented towards narrative reports: Dr Warehouse. J. Biomed. Inform. 2018, 80, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.R.; El-Matary, W.; Valentino, P.L.; Abdou, R.; Alqoaer, K.; Amin, M.; Amir, A.Z.; Auth, M.; Bazerbachi, F.; Broderick, A.; et al. The natural history of primary sclerosing cholangitis in 781 children: A multicenter, international collaboration. Hepatology 2017, 66, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Berry, P.A.; Cross, T.J.; Thein, S.L.; Portmann, B.C.; Wendon, J.A.; Karani, J.B.; Heneghan, M.A.; Bomford, A. Hepatic dysfunction in sickle cell disease: A new system of classification based on global assessment. Clin. Gastroenterol. Hematol. 2007, 5, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Curro, G.; Meo, A.; Ippolito, D.; Pusiol, A.; Cucinetta, E. Asymptomatic cholelithiasis in children with sickle cell disease, early or delayed cholecystectomy? Ann. Surg. 2007, 245, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.F.; Partain, P.I.; Lebensburger, J.D.; Fineberg, N.S.; Howard, T.H. Elective cholecystectomy reduces morbidity of cholelithiasis in pediatric sickle cell disease. Pediatr. Blood Cancer 2017, 64, 113–120. [Google Scholar] [CrossRef]
- Colli, A.; Berzuinin, A.; Prati, D. Liver sequestration in sickle-cell disease and hepatitis. Lancet 2018, 392, e16. [Google Scholar] [CrossRef]
- Howard, J. Sickle cell disease: When and how to transfuse. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 2016, 625–631. [Google Scholar] [CrossRef]
- Li-Thiao-Te, V.; Uettwiller, F.; Quartier, P.; Lacaille, F.; Bader-Meunier, B.; Brousse, V.; De Montalembert, M. Coexistent sickle-cell anemia and autoimmune disease in eight children: Pitfalls and challenges. Pediatr. Rheumatol. Online J. 2018, 16, 5. [Google Scholar] [CrossRef]
- Couillard, S.; Benkerrou, M.; Girot, R.; Brousse, V.; Ferster, A.; Bader-Meunier, B. Steroid treatments in children with sickle cell disease. Haematologica 2007, 92, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, E.; de Vree, J.M.L.; Cresteil, D.; Sokal, E.M.; Sturm, E.; Dumont, M.; Scheffer, G.L.; Paul, M.; Burdelski, M.; Bosma, P.J.; et al. The wide spectrum of Multidrug Resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001, 120, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Issa, H.; Al-Haddad, A.; Al-Salem, A. Sickle cell cholangiopathy: An endoscopic retrograde cholangiopancreatography evaluation. World J. Gastroenterol. 2009, 15, 5316–5320. [Google Scholar] [CrossRef] [PubMed]
- Mekeel, K.L.; Langham, M.R., Jr.; Gonzalez-Peralta, R.; Fujita, S.; Hemming, A.W. Liver transplantation in children with sickle-cell disease. Liver Transpl. 2007, 13, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Hurtova, M.; Bachir, D.; Lee, K.; Calderaro, J.; Descaens, T.; Kluger, M.D.; Zafrani, E.S.; Cherqui, D.; Mallat, A.; Galactéros, F. Transplantation for liver failure in patients with sickle cell disease: Challenging but feasible. Liver Transplant. 2011, 17, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Duvoux, C.; (Paris XII University, Créteil, France). Personal communication, 2019.
- Feld, J.J.; Kato, G.J.; Koh, C.; Shields, T.; Hildesheim, M.; Kleiner, D.E.; Taylor, J.G., 6th; Sandler, N.G.; Douek, D.; Haynes-Williams, V.; et al. Liver injury is associated with mortality in sickle cell disease. Aliment. Pharmacol. Ther. 2015, 42, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Emre, S.; Kitibayashi, K.; Schwartz, M.E.; Ahn, J.; Birnbaum, A.; Thung, S.N.; Miller, C.M. Liver transplantation in a patient with acute liver failure due to sickle cell intrahepatic cholestasis. Transplantation 2000, 69, 675–676. [Google Scholar] [CrossRef] [PubMed]
- De Montalembert, M.; Ribeil, J.A.; Brousse, V.; Guerci-Bresler, A.; Stamatoullas, A.; Vannier, J.P.; Dumesnil, C.; Lahary, A.; Touati, M.; Bouabdallah, K.; et al. Cardiac iron overload in chronically transfused patients with thalassemia, sickle cell anemia, or myelodysplastic syndrome. PLoS ONE 2017, 12, e0172147. [Google Scholar] [CrossRef]
- Darbari, D.S.; Kple-Faget, P.; Kwagyan, J.; Rana, S.; Gordeuk, V.R.; Castro, O. Circumstances of death in adult sickle cell disease patients. Am. J. Hematol. 2006, 81, 858–863. [Google Scholar] [CrossRef]
- Vichinsky, E.; El-Beshlawy, A.; Al Zoebie, A.; Kamden, A.; Koussa, S.; Chotsampancharoen, T.; Bruederle, A.; Gilotti, G.; Han, J.; Elafy, M. Long-term safety and efficacy of deferasirox in young pediatric patients with transfusional hemosiderosis: Results from a 5-year observational study (ENTRUST). Pediatr. Blood Cancer 2017, 64, e26507. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.; Cohen, H. Serious hazards of transfusion (SHOT) haemovigilance and progress is improving transfusion safety. Br. J. Haematol. 2013, 163, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Qhalib, H.A.; Zain, G.H. Hepatobiliary complications of sickle cell disease among children admitted to Al Wahda Teaching Hopsital, Aden, Yemen. Sultan Qaboos Univ. Med. J. 2014, 14, e556–e560. [Google Scholar] [PubMed]
- Zakaria, N.; Knisely, A.; Portmann, B.; Mieili-Vergani, G.; Wendon, J.; Arya, R.; Devlin, J. Acute sickle cell hepatopathy represents a potential contraindication for percutaneous liver biopsy. Blood 2003, 101, 101–103. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lithiasis (Screening or Pain) | Migrating Lithiasis | Cholangio-Pathy | Acute Hepatic Crisis | Acute Hepatic Sequestration | Iron Overload | Suspected Chelator Toxicity | Infectious Hepatitis | Isolated Biochemical Abnormalities | |
|---|---|---|---|---|---|---|---|---|---|
| N | 137 | 19 | 5 | 31 * | 11 | 18 | 5 | 10 | 16 |
| Age (years) | 10.4 (2.5–20; 7.5–12.5) | 10.9 (4.9–19.3; 8.2–13.4) | 9.9 (7.6–14.6; 8.8–11.3) | 12 (1.1–17.6; 6.3–13.8 | 6.3 (0.7–15.4; 4.9–9.6) | 11.5 (4.3–16.9; 8.1–14.5) | 4.9 (3.2–14.3; 4.6–9.9) | 6.8 (2.8–17.6; 4.2–10.6) | 9.9 (2.3–16.9; 8.3–12.2) |
| Female (%) | 69/137 (50%) | 7/19 (37%) | 3/5 (60%) | 13/31 (42%) | 5/11 (45%) | 9/18 (50%) | 1/5 (20%) | 4/10 (40%) | 8/16 (50%) |
| Genotype (SS-S/β0- S/β+-SC) | 124-5- 3-5 | 17-0- 0-2 | 5-0- 0-0 | 28-2- 1-0 | 10-0- 0-1 | 18-0- 0-0 | 5-0- 0-0 | 9-0- 0-1 | 16-0- 0-0 |
| G6PD deficiency (%) | 18/115 (16%) | 3/17 (18%) | 1/4 (25%) | 2/28 (7%) | 4/10 (40%) | 3/13 (23%) | 0/2 (0%) | 0/10 (0%) | 1/12 (8%) |
| VOC (nb/year) | 0.9 ± 1.6 | 1.6 ± 1.6 | 0.2 ± 0.4 | 0.9 ± 1.4 | 0.0 ± 0.0 | 0.6 ± 1.7 | 0.2 ± 0.4 | 0.4 ± 0.7 | 1.6 ± 3.1 |
| HU (%) | 40/137 (29%) | 10/19 (53%) | 4/5 (80%) | 15/31 (48%) | 2/11 (18%) | 2/18 (11%) | 0/5 (0%) | 2/10 (20%) | 7/16 (44%) |
| MET (%) | 19/137 (14%) | 0/19 (0%) | 2/5 (40%) | 6/31 (19%) | 0/11 (0%) | 16/18 (89%) | 5/5 (100%) | 3/10 (30%) | 1/16 (6%) |
| Lithiasis (Screening or Pain) | Migrating Lithiasis | Cholangiopathy | Acute Hepatic Crisis | Acute Hepatic Sequestration | Iron Overload | Suspected Chelator Toxicity | Infectious Hepatitis | Isolated Biochemical Abnormalities | |
|---|---|---|---|---|---|---|---|---|---|
| N | 137 | 19 | 5 | 31 | 11 | 18 | 5 | 10 | 16 |
| Hb (g/dL) | 8.3 (5.1–12.3; 7.3–9.5) | 8.8 (5.5–11.8; 7.1–9.7) | 8.9 (6.6–9.7; 8.4–9.2) | 8.5 (4.1–11.6; 7.2–9.2) | 5.5 (3.9–8.0; 4.9–6.1) | NA | NA | 7.0 (6.3–9.3; 6.5–8.8) | 8.6 (6.3–9.6; 8.0–9.0) |
| ALT (U/L) | 24 (8–210; 17–32) | 105 (17–384; 71–287) | 33 (25–109; 25–96) | 134 (36–1624; 78–297) | 53 (16–213; 47–183) | 25 (13–204; 17–32) | 303 (73–1624; 194–459) | 65 (18–250; 27–238) | 60 (15–255; 36–112) |
| Total bilirubin (µmol/L) | 47 (7–161; 30–70) | 112 (15–375; 56–150) | 50 (33–409; 34–57) | 54 (13–685; 27–118) | 56 (21–570; 32–241) | 47 (20–171; 35–75) | 27 (22–40; 25–31) | 27 (16–52; 22–37) | 52 (12–224; 39–70) |
| Conjugated bilirubin (µmol/L) | 9 (3–63; 8–12) | 37 (6–292; 10–83) | 24 (12–331; 14–31) | 21 (5–465; 11–64) | 20 (7–430; 12–185) | 10 (5–16; 7–10) | 9 (6–30; 7–17) | 11 (6–15; 9–14) | 13 (7–23; 9–15) |
| γ-GT (U/L) | 20 (5–19; 16–30) | 120 (15–432; 53–216) | 115 (19–496; 44–149) | 169 (14–780; 40–264) | 32 (17–162; 20–64) | 17 (12–115; 15–24) | 43 (14–180; 28–86) | 37 (18–143; 35–50) | 50 (15–136; 38–70) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allali, S.; de Montalembert, M.; Brousse, V.; Heilbronner, C.; Taylor, M.; Brice, J.; Manzali, E.; Garcelon, N.; Lacaille, F. Hepatobiliary Complications in Children with Sickle Cell Disease: A Retrospective Review of Medical Records from 616 Patients. J. Clin. Med. 2019, 8, 1481. https://doi.org/10.3390/jcm8091481
Allali S, de Montalembert M, Brousse V, Heilbronner C, Taylor M, Brice J, Manzali E, Garcelon N, Lacaille F. Hepatobiliary Complications in Children with Sickle Cell Disease: A Retrospective Review of Medical Records from 616 Patients. Journal of Clinical Medicine. 2019; 8(9):1481. https://doi.org/10.3390/jcm8091481
Chicago/Turabian StyleAllali, Slimane, Mariane de Montalembert, Valentine Brousse, Claire Heilbronner, Melissa Taylor, Josephine Brice, Elisabetta Manzali, Nicolas Garcelon, and Florence Lacaille. 2019. "Hepatobiliary Complications in Children with Sickle Cell Disease: A Retrospective Review of Medical Records from 616 Patients" Journal of Clinical Medicine 8, no. 9: 1481. https://doi.org/10.3390/jcm8091481
APA StyleAllali, S., de Montalembert, M., Brousse, V., Heilbronner, C., Taylor, M., Brice, J., Manzali, E., Garcelon, N., & Lacaille, F. (2019). Hepatobiliary Complications in Children with Sickle Cell Disease: A Retrospective Review of Medical Records from 616 Patients. Journal of Clinical Medicine, 8(9), 1481. https://doi.org/10.3390/jcm8091481