Bone Marrow-Derived Mesenchymal Stromal Cells: A Novel Target to Optimize Hematopoietic Stem Cell Transplantation Protocols in Hematological Malignancies and Rare Genetic Disorders

 ,
,
Abstract
: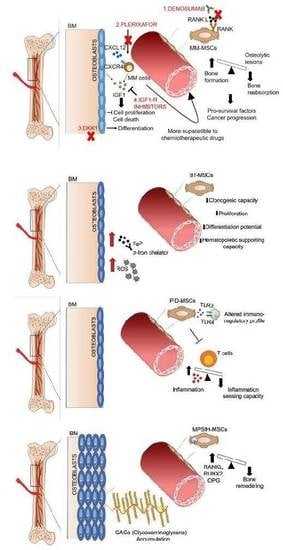
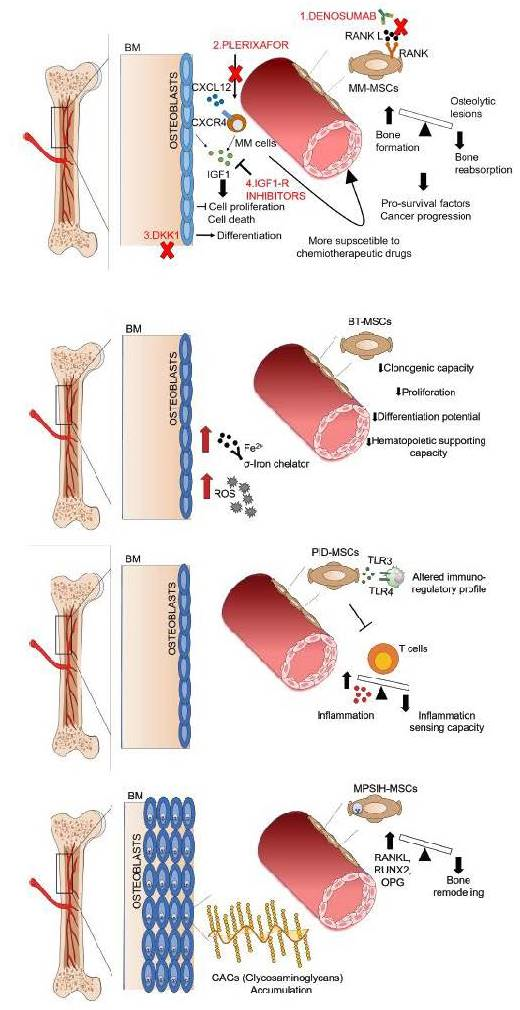{kind=link}
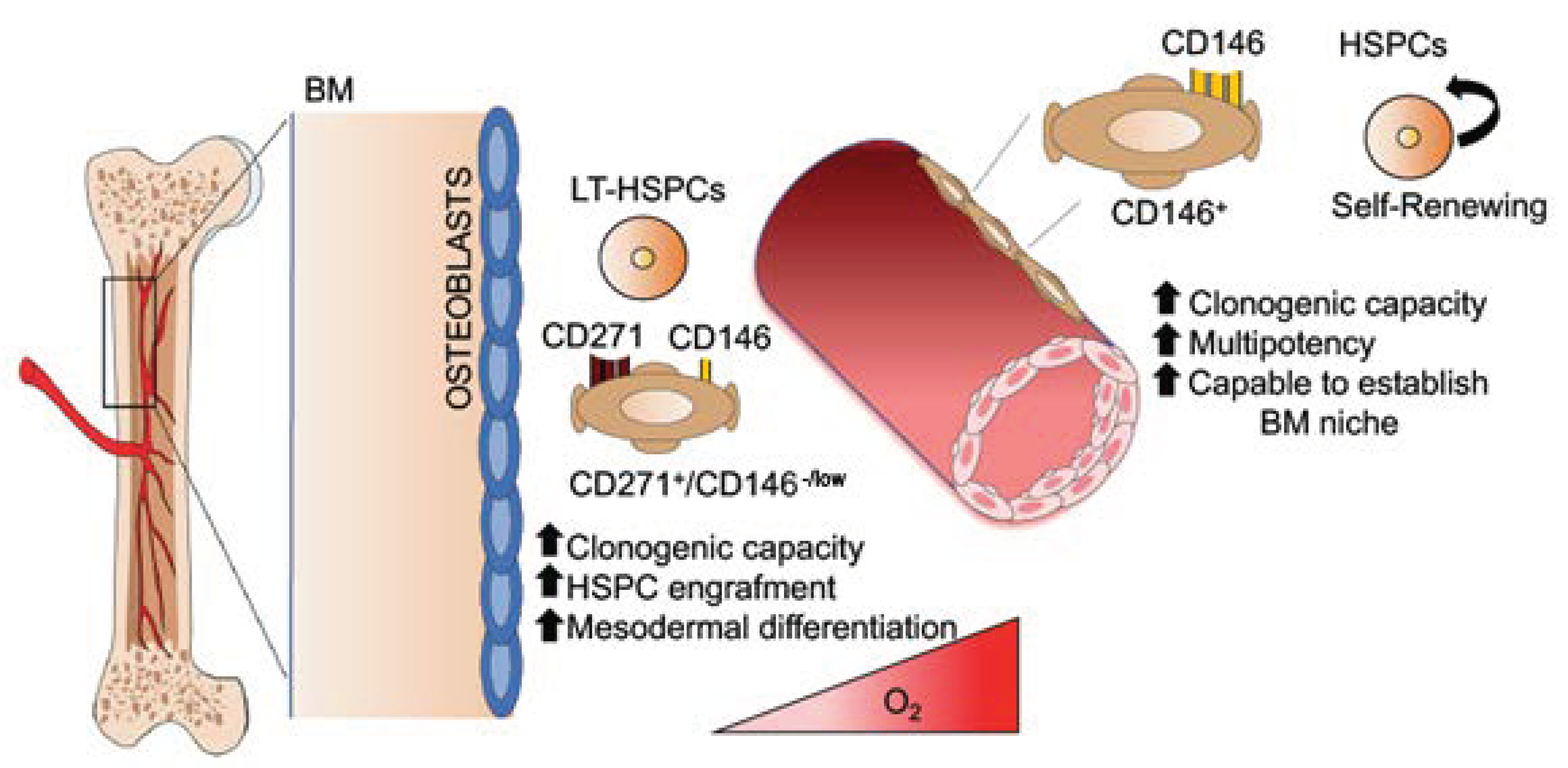{kind=link}
{kind=link}
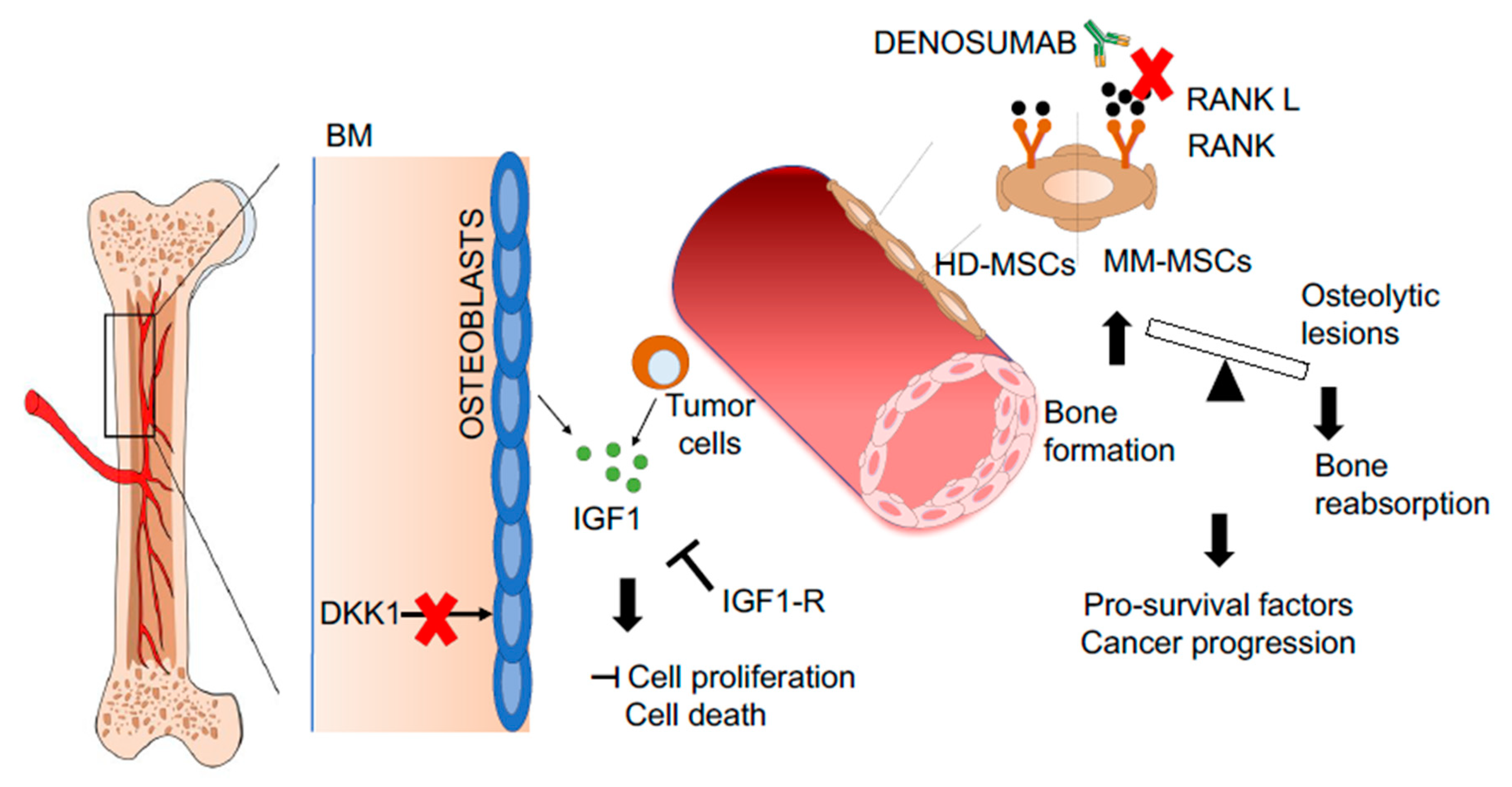{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MSCs as Key Elements of the Bone Marrow Niche
3. MSCs in the Clinical Use
4. MSCs in Hematological Malignancies
5. Targeting BM Stroma: A Novel Therapeutic Approach to Treat Hematological Malignancies
6. MSCs in Rare Genetic Diseases
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet 1970, 3, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.J.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Mendes, S.C.; Robin, C.; Dzierzak, E. Mesenchymal progenitor cells localize within hematopoietic sites throughout ontogeny. Development 2005, 132, 1127–1136. [Google Scholar] [CrossRef] [Green Version]
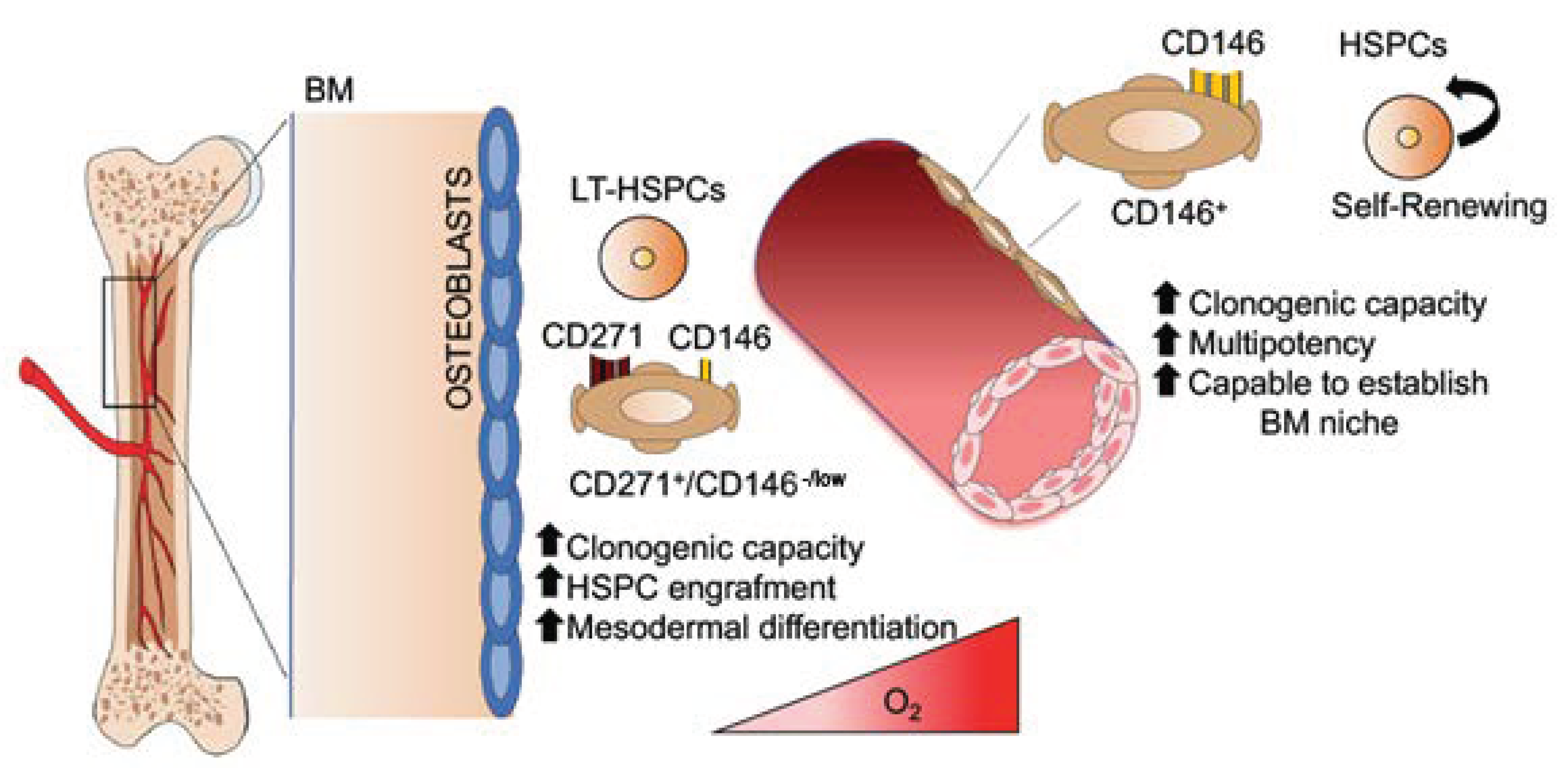
- Quirici, N.; Soligo, D.; Bossolasco, P.; Servida, F.; Lumini, C.; Deliliers, G.L. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exp. Hematol. 2002, 30, 783–791. [Google Scholar] [CrossRef]
- Churchman, S.M.; Ponchel, F.; Boxall, S.A.; Cuthbert, R.; Kouroupis, D.; Roshdy, T.; Giannoudis, P.V.; Emery, P.; McGonagle, D.; Jones, E.A. Transcriptional profile of native CD271+ multipotential stromal cells: Evidence for multiple fates, with prominent osteogenic and Wnt pathway signaling activity. Arthritis Rheum. 2012, 64, 2632–2643. [Google Scholar] [CrossRef]
- Battula, V.L.; Treml, S.; Bareiss, P.M.; Gieseke, F.; Roelofs, H.; de Zwart, P.; Muller, I.; Schewe, B.; Skutella, T.; Fibbe, W.E.; et al. Isolation of functionally distinct mesenchymal stem cell subsets using antibodies against CD56, CD271, and mesenchymal stem cell antigen-1. Haematologica 2009, 94, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormin, A.; Li, O.; Brune, J.C.; Walsh, S.; Schutz, B.; Ehinger, M.; Ditzel, N.; Kassem, M.; Scheding, S. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 2011, 117, 5067–5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ghazanfari, R.; Zacharaki, D.; Ditzel, N.; Isern, J.; Ekblom, M.; Mendez-Ferrer, S.; Kassem, M.; Scheding, S. Low/negative expression of PDGFR-alpha identifies the candidate primary mesenchymal stromal cells in adult human bone marrow. Stem Cell Rep. 2014, 3, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, Y.; Morikawa, S.; Harada, S.; Niibe, K.; Suzuki, S.; Renault-Mihara, F.; Houlihan, D.D.; Akazawa, C.; Okano, H.; Matsuzaki, Y. LNGFR(+)THY-1(+)VCAM-1(hi+) cells reveal functionally distinct subpopulations in mesenchymal stem cells. Stem Cell Rep. 2013, 1, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.; Lacombe, J.; Hanoun, M.; Mizoguchi, T.; Bruns, I.; Kunisaki, Y.; Frenette, P.S. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013, 210, 1351–1367. [Google Scholar] [CrossRef]
- Jones, E.A.; Kinsey, S.E.; English, A.; Jones, R.A.; Straszynski, L.; Meredith, D.M.; Markham, A.F.; Jack, A.; Emery, P.; McGonagle, D. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002, 46, 3349–3360. [Google Scholar] [CrossRef]
- Kuci, S.; Kuci, Z.; Kreyenberg, H.; Deak, E.; Putsch, K.; Huenecke, S.; Amara, C.; Koller, S.; Rettinger, E.; Grez, M.; et al. CD271 antigen defines a subset of multipotent stromal cells with immunosuppressive and lymphohematopoietic engraftment-promoting properties. Haematologica 2010, 95, 651–659. [Google Scholar] [CrossRef]
- Ganguly, P.; El-Jawhari, J.J.; Burska, A.N.; Ponchel, F.; Giannoudis, P.V.; Jones, E.A. The analysis of in vivo aging in human bone marrow mesenchymal stromal cells using colony-forming unit-fibroblast assay and the CD45(low)CD271(+) phenotype. Stem Cells Int. 2019, 2019, 5197983. [Google Scholar] [CrossRef] [Green Version]
- Secunda, R.; Vennila, R.; Mohanashankar, A.M.; Rajasundari, M.; Jeswanth, S.; Surendran, R. Isolation, expansion and characterisation of mesenchymal stem cells from human bone marrow, adipose tissue, umbilical cord blood and matrix: A comparative study. Cytotechnology 2015, 67, 793–807. [Google Scholar] [CrossRef]
- Beeravolu, N.; McKee, C.; Alamri, A.; Mikhael, S.; Brown, C.; Perez-Cruet, M.; Chaudhry, G.R. Isolation and characterization of mesenchymal stromal cells from human umbilical cord and fetal placenta. J. Vis. Exp. 2017, 122, e55224. [Google Scholar] [CrossRef] [Green Version]
- Arana, M.; Mazo, M.; Aranda, P.; Pelacho, B.; Prosper, F. Adipose tissue-derived mesenchymal stem cells: Isolation, expansion, and characterization. Methods Mol. Biol. 2013, 1036, 47–61. [Google Scholar] [PubMed]
- Ghazanfari, R.; Zacharaki, D.; Li, H.; Ching Lim, H.; Soneji, S.; Scheding, S. Human primary bone marrow mesenchymal stromal cells and their in vitro progenies display distinct transcriptional profile signatures. Sci. Rep. 2017, 7, 10338. [Google Scholar] [CrossRef] [PubMed]
- Mosna, F.; Sensebe, L.; Krampera, M. Human bone marrow and adipose tissue mesenchymal stem cells: A user’s guide. Stem Cells Dev. 2010, 19, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Colter, D.C.; Sekiya, I.; Prockop, D.J. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7841–7845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selich, A.; Ha, T.C.; Morgan, M.; Falk, C.S.; von Kaisenberg, C.; Schambach, A.; Rothe, M. Cytokine selection of MSC clones with different functionality. Stem Cell Rep. 2019, 13, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Abbuehl, J.P.; Tatarova, Z.; Held, W.; Huelsken, J. Long-term engraftment of primary bone marrow stromal cells repairs niche damage and improves hematopoietic stem cell transplantation. Cell Stem Cell 2017, 21, 241–255. [Google Scholar] [CrossRef]
- Zhou, Y.; Tsai, T.L.; Li, W.J. Strategies to retain properties of bone marrow-derived mesenchymal stem cells ex vivo. Ann. N. Y. Acad. Sci. 2017, 1409, 3–17. [Google Scholar] [CrossRef]
- Li, Y.M.; Schilling, T.; Benisch, P.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Limbert, C.; Seufert, J.; Kassem, M.; Schutze, N.; et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem. Biophys Res. Commun. 2007, 363, 209–215. [Google Scholar] [CrossRef]
- Lv, H.; Li, L.; Sun, M.; Zhang, Y.; Chen, L.; Rong, Y.; Li, Y. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res. Ther. 2015, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, F.; Borger, D.K.; Wei, Q.; Pinho, S.; Maryanovich, M.; Zahalka, A.H.; Suzuki, M.; Cruz, C.D.; Wang, Z.; Xu, C.; et al. Engineering a haematopoietic stem cell niche by revitalizing mesenchymal stromal cells. Nat. Cell Biol. 2019, 21, 560–567. [Google Scholar] [CrossRef]
- Rohban, R.; Pieber, T.R. Mesenchymal stem and progenitor cells in regeneration: Tissue specificity and regenerative potential. Stem Cells Int. 2017, 2017, 5173732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinert, A.F.; Rackwitz, L.; Gilbert, F.; Noth, U.; Tuan, R.S. Concise review: The clinical application of mesenchymal stem cells for musculoskeletal regeneration: Current status and perspectives. Stem Cells Transl. Med. 2012, 1, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Galipeau, J.; Kessler, C.; le Blanc, K.; Dazzi, F. Challenges for mesenchymal stromal cell therapies. Sci. Transl. Med. 2019, 11, eaat2189. [Google Scholar] [CrossRef] [PubMed]
- le Blanc, K.; Davies, L.C. Mesenchymal stromal cells and the innate immune response. Immunol. Lett. 2015, 168, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauta, A.J.; Fibbe, W.E. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007, 110, 3499–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Krampera, M. Mesenchymal stromal cell ‘licensing’: A multistep process. Leukemia 2011, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Galipeau, J. Mechanisms of immune modulation by mesenchymal stromal cells and clinical translation. Curr. Mol. Med. 2013, 13, 856–867. [Google Scholar] [CrossRef]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, M.E.; Ball, L.M.; Cometa, A.M.; Roelofs, H.; Zecca, M.; Avanzini, M.A.; Bertaina, A.; Vinti, L.; Lankester, A.; Maccario, R.; et al. Co-infusion of ex vivo-expanded, parental MSCs prevents life-threatening acute GVHD, but does not reduce the risk of graft failure in pediatric patients undergoing allogeneic umbilical cord blood transplantation. Bone Marrow Transplant. 2011, 46, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Ball, L.M.; Bernardo, M.E.; Roelofs, H.; Lankester, A.; Cometa, A.; Egeler, R.M.; Locatelli, F.; Fibbe, W.E. Cotransplantation of ex vivo expanded mesenchymal stem cells accelerates lymphocyte recovery and may reduce the risk of graft failure in haploidentical hematopoietic stem-cell transplantation. Blood 2007, 110, 2764–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.N.; Ng, J.; Niu, T.; Yang, H.; McMannis, J.D.; Karandish, S.; Kaur, I.; Fu, P.; Del Angel, M.; Messinger, R.; et al. Superior ex vivo cord blood expansion following co-culture with bone marrow-derived mesenchymal stem cells. Bone Marrow Transplant. 2006, 37, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lima, M.; McNiece, I.; Robinson, S.N.; Munsell, M.; Eapen, M.; Horowitz, M.; Alousi, A.; Saliba, R.; McMannis, J.D.; Kaur, I.; et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N. Engl. J. Med. 2012, 367, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najar, M.; Bouhtit, F.; Melki, R.; Afif, H.; Hamal, A.; Fahmi, H.; Merimi, M.; Lagneaux, L. Mesenchymal stromal cell-based therapy: New perspectives and challenges. J. Clin. Med. 2019, 8, E626. [Google Scholar] [CrossRef] [Green Version]
- Birbrair, A.; Frenette, P.S. Niche heterogeneity in the bone marrow. Ann. N. Y. Acad. Sci. 2016, 1370, 82–96. [Google Scholar] [CrossRef]
- Szade, K.; Gulati, G.S.; Chan, C.K.F.; Kao, K.S.; Miyanishi, M.; Marjon, K.D.; Sinha, R.; George, B.M.; Chen, J.Y.; Weissman, I.L. Where hematopoietic stem cells live: The bone marrow niche. Antioxid. Redox Signal. 2018, 29, 191–204. [Google Scholar] [CrossRef]
- Asada, N.; Takeishi, S.; Frenette, P.S. Complexity of bone marrow hematopoietic stem cell niche. Int. J. Hematol. 2017, 106, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, M.K.; Thiede, M.A.; Haynesworth, S.E.; Bruder, S.P.; Gerson, S.L. Human marrow-derived mesenchymal stem cells (MSCs) express hematopoietic cytokines and support long-term hematopoiesis when differentiated toward stromal and osteogenic lineages. J. Hematother. Stem Cell Res. 2000, 9, 841–848. [Google Scholar] [CrossRef]
- Osugi, M.; Katagiri, W.; Yoshimi, R.; Inukai, T.; Hibi, H.; Ueda, M. Conditioned media from mesenchymal stem cells enhanced bone regeneration in rat calvarial bone defects. Tissue Eng. Part A 2012, 18, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Lampreia, F.P.; Carmelo, J.G.; Anjos-Afonso, F. Notch signaling in the regulation of hematopoietic stem cell. Curr. Stem Cell Rep. 2017, 3, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Corselli, M.; Crisan, M.; Murray, I.R.; West, C.C.; Scholes, J.; Codrea, F.; Khan, N.; Peault, B. Identification of perivascular mesenchymal stromal/stem cells by flow cytometry. Cytom. A 2013, 83, 714–720. [Google Scholar] [CrossRef]
- Boulais, M.; Soudant, P.; Le Goic, N.; Quere, C.; Boudry, P.; Suquet, M. Involvement of mitochondrial activity and OXPHOS in ATP synthesis during the motility phase of spermatozoa in the pacific oyster, crassostrea gigas. Biol. Reprod. 2015, 93, 118. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisan, M.; Huard, J.; Zheng, B.; Sun, B.; Yap, S.; Logar, A.; Giacobino, J.P.; Casteilla, L.; Peault, B. Purification and culture of human blood vessel-associated progenitor cells. Curr. Protoc. Stem Cell Biol. 2008, 4, 2B.2.1–2B.2.13. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arinobu, Y.; Mizuno, S.; Chong, Y.; Shigematsu, H.; Iino, T.; Iwasaki, H.; Graf, T.; Mayfield, R.; Chan, S.; Kastner, P.; et al. Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell 2007, 1, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- Sorrentino, A.; Ferracin, M.; Castelli, G.; Biffoni, M.; Tomaselli, G.; Baiocchi, M.; Fatica, A.; Negrini, M.; Peschle, C.; Valtieri, M. Isolation and characterization of CD146+ multipotent mesenchymal stromal cells. Exp. Hematol. 2008, 36, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.C.; Phinney, D.G.; Lacey, M.R.; Barrilleaux, B.L.; Meyertholen, K.E.; O’Connor, K.C. In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells 2010, 28, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Simmons, P.J.; Torok-Storb, B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood 1991, 78, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensidhoum, M.; Chapel, A.; Francois, S.; Demarquay, C.; Mazurier, C.; Fouillard, L.; Bouchet, S.; Bertho, J.M.; Gourmelon, P.; Aigueperse, J.; et al. Homing of in vitro expanded Stro-1- or Stro-1+ human mesenchymal stem cells into the NOD/SCID mouse and their role in supporting human CD34 cell engraftment. Blood 2004, 103, 3313–3319. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Bosnakovski, D.; Figueiredo, C.A.; Visser, J.W.; Perlingeiro, R.C. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood 2007, 109, 1743–1751. [Google Scholar] [CrossRef]
- Suila, H.; Pitkanen, V.; Hirvonen, T.; Heiskanen, A.; Anderson, H.; Laitinen, A.; Natunen, S.; Miller-Podraza, H.; Satomaa, T.; Natunen, J.; et al. Are globoseries glycosphingolipids SSEA-3 and -4 markers for stem cells derived from human umbilical cord blood? J. Mol. Cell Biol. 2011, 3, 99–107. [Google Scholar] [CrossRef]
- Reinisch, A.; Hernandez, D.C.; Schallmoser, K.; Majeti, R. Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat. Protoc. 2017, 12, 2169–2188. [Google Scholar] [CrossRef]
- Abarrategi, A.; Mian, S.A.; Passaro, D.; Rouault-Pierre, K.; Grey, W.; Bonnet, D. Modeling the human bone marrow niche in mice: From host bone marrow engraftment to bioengineering approaches. J. Exp. Med. 2018, 215, 729–743. [Google Scholar] [CrossRef] [Green Version]
- Krampera, M.; Galipeau, J.; Shi, Y.; Tarte, K.; Sensebe, L.; M. S. C. Committee of the International Society for Cellular Therapy. Immunological characterization of multipotent mesenchymal stromal cells—The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy 2013, 15, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Meisel, R.; Zibert, A.; Laryea, M.; Gobel, U.; Daubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uccelli, A.; Moretta, L.; Pistoia, V. Immunoregulatory function of mesenchymal stem cells. Eur. J. Immunol. 2006, 36, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Frassoni, F.; Ball, L.; Locatelli, F.; Roelofs, H.; Lewis, I.; Lanino, E.; Sundberg, B.; Bernardo, M.E.; Remberger, M.; et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase II study. Lancet 2008, 371, 1579–1586. [Google Scholar] [CrossRef]
- Zhou, Y.; Yamamoto, Y.; Xiao, Z.; Ochiya, T. The immunomodulatory functions of mesenchymal stromal/stem cells mediated via paracrine activity. J. Clin. Med. 2019, 8, 1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, C.; Ratajczak, M.Z.; Laughlin, M.J. Strategies to enhance umbilical cord blood stem cell engraftment in adult patients. Expert Rev. Hematol. 2010, 3, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Weng, J.Y.; Du, X.; Geng, S.X.; Peng, Y.W.; Wang, Z.; Lu, Z.S.; Wu, S.J.; Luo, C.W.; Guo, R.; Ling, W.; et al. Mesenchymal stem cell as salvage treatment for refractory chronic GVHD. Bone Marrow Transplant. 2010, 45, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, M.; Abbaszadeh, H.; Movassaghpour, A.A.; Mehdizadeh, A.; Ebrahimi-Warkiani, M.; Yousefi, M. Strategies for elevating hematopoietic stem cells expansion and engraftment capacity. Life Sci. 2019, 232, 116598. [Google Scholar] [CrossRef]
- Noort, W.A.; Kruisselbrink, A.B.; In’t Anker, P.S.; Kruger, M.; van Bezooijen, R.L.; de Paus, R.A.; Heemskerk, M.H.; Lowik, C.W.; Falkenburg, J.H.; Willemze, R.; et al. Mesenchymal stem cells promote engraftment of human umbilical cord blood-derived CD34(+) cells in NOD/SCID mice. Exp. Hematol. 2002, 30, 870–878. [Google Scholar] [CrossRef]
- Almeida-Porada, G.; Porada, C.D.; Tran, N.; Zanjani, E.D. Cotransplantation of human stromal cell progenitors into preimmune fetal sheep results in early appearance of human donor cells in circulation and boosts cell levels in bone marrow at later time points after transplantation. Blood 2000, 95, 3620–3627. [Google Scholar] [CrossRef]
- Masuda, S.; Ageyama, N.; Shibata, H.; Obara, Y.; Ikeda, T.; Takeuchi, K.; Ueda, Y.; Ozawa, K.; Hanazono, Y. Cotransplantation with MSCs improves engraftment of HSCs after autologous intra-bone marrow transplantation in nonhuman primates. Exp. Hematol. 2009, 37, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, N.; Cho, S.G. The potential use of mesenchymal stem cells in hematopoietic stem cell transplantation. Exp. Mol. Med. 2013, 45, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, W.; Roderburg, C.; Wein, F.; Diehlmann, A.; Frankhauser, M.; Schubert, R.; Eckstein, V.; Ho, A.D. Molecular and secretory profiles of human mesenchymal stromal cells and their abilities to maintain primitive hematopoietic progenitors. Stem Cells 2007, 25, 2638–2647. [Google Scholar] [CrossRef] [PubMed]
- Kadekar, D.; Kale, V.; Limaye, L. Differential ability of MSCs isolated from placenta and cord as feeders for supporting ex vivo expansion of umbilical cord blood derived CD34(+) cells. Stem Cell Res. Ther. 2015, 6, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, C.; Tonnarelli, B.; Papadimitropoulos, A.; Scherberich, A.; Schaeren, S.; Schauerte, A.; Lopez-Rios, J.; Zeller, R.; Barbero, A.; Martin, I. Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 7251–7256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Occhetta, P.; Pigeot, S.; Rasponi, M.; Dasen, B.; Mehrkens, A.; Ullrich, T.; Kramer, I.; Guth-Gundel, S.; Barbero, A.; Martin, I. Developmentally inspired programming of adult human mesenchymal stromal cells toward stable chondrogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4625–4630. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.M.; Walker, R.V.; Parker, S.; Rhodes, N.P.; Hunt, J.A.; Freemont, A.J.; Hoyland, J.A. Intervertebral disc cell-mediated mesenchymal stem cell differentiation. Stem Cells 2006, 24, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Rajabzadeh, N.; Fathi, E.; Farahzadi, R. Stem cell-based regenerative medicine. Stem Cell Investig. 2019, 6, 19. [Google Scholar] [CrossRef]
- Gunawardena, T.N.A.; Rahman, M.T.; Abdullah, B.J.J.; Kasim, N.H.A. Conditioned media derived from mesenchymal stem cell cultures: The next generation for regenerative medicine. J. Tissue Eng. Regen. Med. 2019, 13, 569–586. [Google Scholar] [CrossRef]
- Venkatesh, K.; Sen, D. Mesenchymal stem cells as a source of dopaminergic neurons: A potential cell based therapy for parkinson’s disease. Curr. Stem Cell Res. Ther. 2017, 12, 326–347. [Google Scholar] [CrossRef]
- Shichinohe, H.; Kawabori, M.; Iijima, H.; Teramoto, T.; Abumiya, T.; Nakayama, N.; Kazumata, K.; Terasaka, S.; Arato, T.; Houkin, K. Research on advanced intervention using novel bone marrOW stem cell (RAINBOW): A study protocol for a phase I, open-label, uncontrolled, dose-response trial of autologous bone marrow stromal cell transplantation in patients with acute ischemic stroke. BMC Neurol. 2017, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Stark, J.; Vyshkina, T.; Blackshear, L.; Joo, G.; Stefanova, V.; Sara, G.; Sadiq, S.A. Phase I trial of intrathecal mesenchymal stem cell-derived neural progenitors in progressive multiple sclerosis. EBioMedicine 2018, 29, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noronha-Matos, J.B.; Correia-de-Sa, P. Mesenchymal stem cells ageing: Targeting the “purinome” to promote osteogenic differentiation and bone repair. J. Cell Physiol. 2016, 231, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Samsonraj, R.M.; Raghunath, M.; Nurcombe, V.; Hui, J.H.; van Wijnen, A.J.; Cool, S.M. Concise review: Multifaceted characterization of human mesenchymal stem cells for use in regenerative medicine. Stem Cells Transl. Med. 2017, 6, 2173–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahsoun, S.; Coopman, K.; Forsyth, N.R.; Akam, E.C. The role of dissolved oxygen levels on human mesenchymal stem cell culture success, regulatory compliance, and therapeutic potential. Stem Cells Dev. 2018, 27, 1303–1321. [Google Scholar] [CrossRef] [Green Version]
- Corradi, G.; Baldazzi, C.; Ocadlikova, D.; Marconi, G.; Parisi, S.; Testoni, N.; Finelli, C.; Cavo, M.; Curti, A.; Ciciarello, M. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res. Ther. 2018, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2019, 1–13. [Google Scholar] [CrossRef]
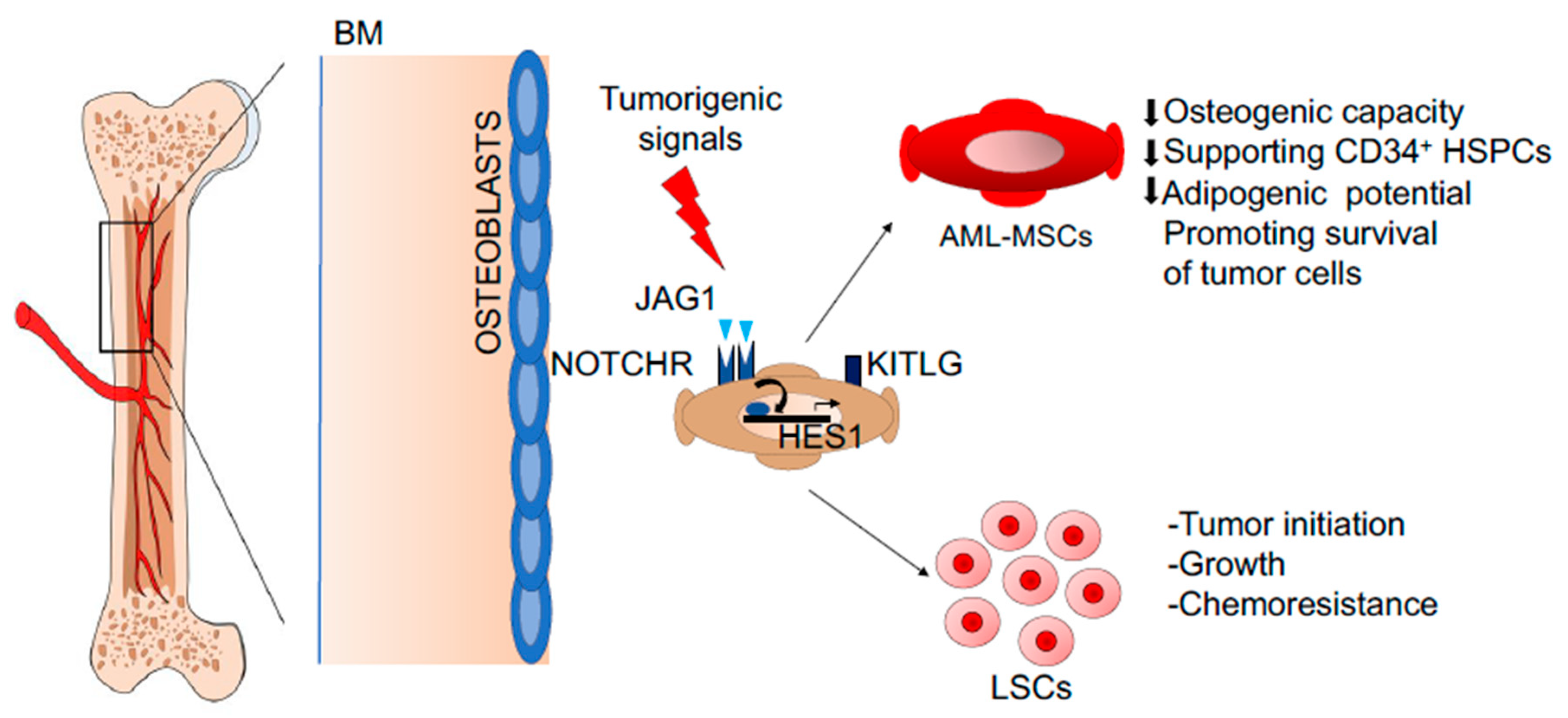
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef]
- Desbourdes, L.; Javary, J.; Charbonnier, T.; Ishac, N.; Bourgeais, J.; Iltis, A.; Chomel, J.C.; Turhan, A.; Guilloton, F.; Tarte, K.; et al. Alteration analysis of bone marrow mesenchymal stromal cells from de novo acute myeloid leukemia patients at diagnosis. Stem Cells Dev. 2017, 26, 709–722. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Kamga, P.T.; Dal Collo, G.; Bassi, G.; Midolo, M.; Delledonne, M.; Chilosi, M.; Bonifacio, M.; Krampera, M. Characterization of a new B-ALL cell line with constitutional defect of the Notch signaling pathway. Oncotarget 2018, 9, 18341–18350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castano, J.; Gomez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed characterization of mesenchymal stem/stromal cells from a large cohort of AML patients demonstrates a definitive link to treatment outcomes. Stem Cell Rep. 2017, 8, 1573–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.K.; Nepstad, I.; Bruserud, O. Mesenchymal stem cells support survival and proliferation of primary human acute myeloid leukemia cells through heterogeneous molecular mechanisms. Front. Immunol. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnani, D.; Crippa, S.; Della Volpe, L.; Rossella, V.; Conti, A.; Lettera, E.; Rivis, S.; Ometti, M.; Fraschini, G.; Bernardo, M.E.; et al. An early-senescence state in aged mesenchymal stromal cells contributes to hematopoietic stem and progenitor cell clonogenic impairment through the activation of a pro-inflammatory program. Aging Cell 2019, 18, e12933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, W.; Horn, P.; Castoldi, M.; Diehlmann, A.; Bork, S.; Saffrich, R.; Benes, V.; Blake, J.; Pfister, S.; Eckstein, V.; et al. Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS ONE 2008, 3, e2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde, M.C.; Demaria, M.; Campisi, J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip. Top Gerontol. 2013, 38, 17–27. [Google Scholar]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Fröbel, J.; Brückner, B.; Kündgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Mattiucci, D.; Maurizi, G.; Leoni, P.; Poloni, A. Aging- and senescence-associated changes of mesenchymal stromal cells in myelodysplastic syndromes. Cell Transplant. 2018, 27, 754–764. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan-Wong, K.; Nadeau, S.; Carrier-Leclerc, A.; Apablaza, F.; Hamdy, R.; Shum-Tim, D.; Rodier, F.; Colmegna, I. Increased IL-6 secretion by aged human mesenchymal stromal cells disrupts hematopoietic stem and progenitor cells’ homeostasis. Oncotarget 2016, 7, 13285–13296. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, J.; Fei, C.; Zheng, Q.; Li, X.; Chang, C. Downregulation of MMP1 in MDS-derived mesenchymal stromal cells reduces the capacity to restrict MDS cell proliferation. Sci. Rep. 2017, 7, 43849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeg, H.J. Hematopoietic cell transplantation for myelodysplastic syndrome. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e375–e380. [Google Scholar] [CrossRef] [PubMed]
- Klaus, M.; Stavroulaki, E.; Kastrinaki, M.C.; Fragioudaki, P.; Giannikou, K.; Psyllaki, M.; Pontikoglou, C.; Tsoukatou, D.; Mamalaki, C.; Papadaki, H.A. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem Cells Dev. 2010, 19, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Fracchiolla, N.S.; Fattizzo, B.; Cortelezzi, A. Mesenchymal stem cells in myeloid malignancies: A focus on immune escaping and therapeutic implications. Stem Cells Int. 2017, 2017, 6720594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.G.; Xu, W.; Yu, H.P.; Fang, B.L.; Wu, S.H.; Li, F.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer Lett. 2012, 317, 136–143. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, X.; Xu, W.; Cao, Z.; Sun, L.; Li, W.; Li, Q.; Zou, P.; Zhao, Z. The different immunoregulatory functions on dendritic cells between mesenchymal stem cells derived from bone marrow of patients with low-risk or high-risk myelodysplastic syndromes. PLoS ONE 2013, 8, e57470. [Google Scholar] [CrossRef]
- Ciciarello, M.; Corradi, G.; Loscocco, F.; Visani, G.; Monaco, F.; Cavo, M.; Curti, A.; Isidori, A. The yin and yang of the bone marrow microenvironment: pros and cons of mesenchymal stromal cells in acute myeloid leukemia. Front. Oncol. 2019, 9, 1135. [Google Scholar] [CrossRef] [Green Version]
- Vicente López, Á.; Vázquez García, M.N.; Melen, G.J.; Entrena Martínez, A.; Cubillo Moreno, I.; García-Castro, J.; Orellana, M.R.; González, A.G. Mesenchymal stromal cells derived from the bone marrow of acute lymphoblastic leukemia patients show altered BMP4 production: Correlations with the course of disease. PLoS ONE 2014, 9, e84496. [Google Scholar] [CrossRef] [Green Version]
- Vilchis-Ordoñez, A.; Contreras-Quiroz, A.; Vadillo, E.; Dorantes-Acosta, E.; Reyes-López, A.; Quintela-Nuñez del Prado, H.M.; Venegas-Vázquez, J.; Mayani, H.; Ortiz-Navarrete, V.; López-Martínez, B.; et al. Bone marrow cells in acute lymphoblastic leukemia create a proinflammatory microenvironment influencing normal hematopoietic differentiation fates. BioMed Res. Int. 2015, 2015, 386165. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukaemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaría, C.; Muntión, S.; Rosón, B.; Blanco, B.; López-Villar, O.; Carrancio, S.; Sánchez-Guijo, F.M.; Díez-Campelo, M.; Alvarez-Fernández, S.; Sarasquete, M.E.; et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 2012, 97, 1218–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupec, R.A.; Jundt, F.; Rebholz, B.; Eckelt, B.; Weindl, G.; Herzinger, T.; Flaig, M.J.; Moosmann, S.; Plewig, G.; Dörken, B.; et al. Stroma-mediated dysregulation of myelopoiesis in mice lacking I kappa B alpa. Immunity 2005, 22, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohda, S.; Nara, N. Expression of Notch1 and Jagged1 proteins in acute myeloid leukemia cells. Leuk. Lymphoma 2001, 42, 467–472. [Google Scholar] [CrossRef]
- Walkley, C.R.; Shea, J.M.; Sims, N.A.; Purton, L.E.; Orkin, S.H. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 2007, 129, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Zandecki, M.; Facon, T.; Preudhomme, C.; Vanrumbeke, M.; Vachee, A.; Quesnel, B.; Lai, J.L.; Cosson, A.; Fenaux, P. The retinoblastoma gene (RB-1) status in multiple myeloma: A report on 35 cases. Leuk. Lymphoma 1995, 18, 497–503. [Google Scholar] [CrossRef]
- Hideshima, T.; Anderson, K.C. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat. Rev. Cancer 2002, 2, 927–937. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: Interplay of growth factors, their receptors and stromal interactions. Eur. J. Cancer 2006, 42, 1564–1573. [Google Scholar] [CrossRef]
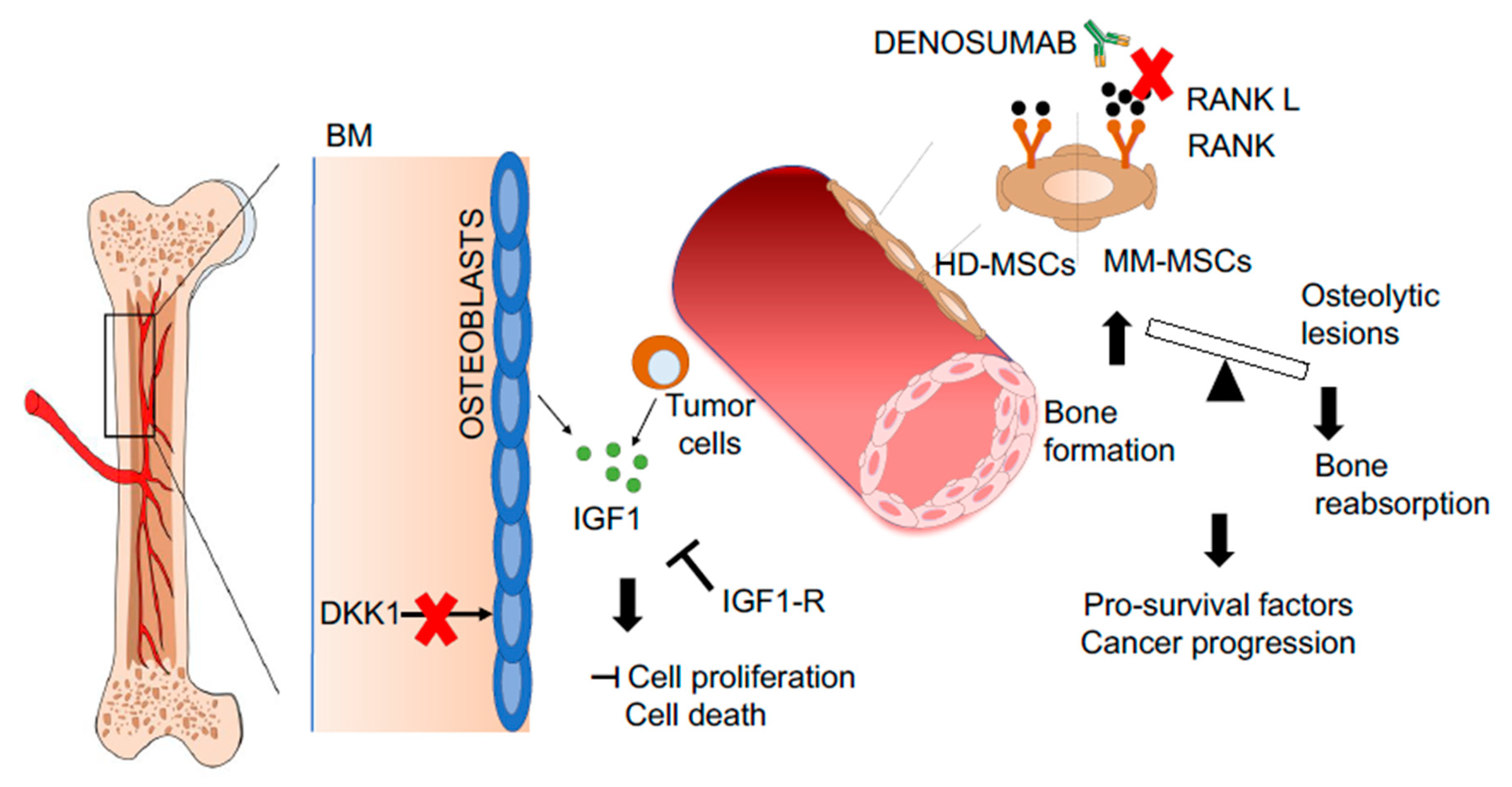
- Giuliani, N.; Colla, S.; Sala, R.; Moroni, M.; Lazzaretti, M.; La Monica, S.; Bonomini, S.; Hojden, M.; Sammarelli, G.; Barillè, S.; et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood 2002, 100, 4615–4621. [Google Scholar] [CrossRef] [Green Version]
- Pearse, R.N.; Sordillo, E.M.; Yaccoby, S.; Wong, B.R.; Liau, D.F.; Colman, N.; Michaeli, J.; Epstein, J.; Choi, Y. Multiple myeloma disrupts the TRANCE/ osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc. Natl. Acad. Sci. USA 2001, 98, 11581–11586. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, E.M.; Ritchlin, C.T. Clinical development of anti-RANKL therapy. Arthritis Res. Ther. 2007, 9 (Suppl. 1), S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, E.S.; Lawson, M.A.; Snowden, J.A.; Jolley, I.; Chantry, A.D. New agents in the treatment of myeloma bone disease. Calcif. Tissue Int. 2018, 102, 196–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, D.A. Denosumab for bone lesions in multiple myeloma-what is its value? Haematologica 2018, 103, 753–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, Y.W.; Chen, Y.; Stephens, O.; Brown, N.; Chen, B.; Epstein, J.; Barlogie, B.; Shaughnessy, J.D. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: A potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 2008, 112, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Pinzone, J.J.; Hall, B.M.; Thudi, N.K.; Vonau, M.; Qiang, Y.W.; Rosol, T.J.; Shaughnessy, J.D. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 2009, 113, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Borrello, I. Can we change the disease biology of multiple myeloma? Leuk. Res. 2012, 36 (Suppl. 1), S3–S12. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Tarte, K.; Legouffe, E.; Brochier, J.; Rossi, J.F.; Klein, B. Tumor necrosis factor is a survival and proliferation factor for human myeloma cells. Eur. Cytokine Netw. 1999, 10, 65–70. [Google Scholar]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [Green Version]
- Malfuson, J.V.; Boutin, L.; Clay, D.; Thépenier, C.; Desterke, C.; Torossian, F.; Guerton, B.; Anginot, A.; de Revel, T.; Lataillade, J.J.; et al. SP/drug efflux functionality of hematopoietic progenitors is controlled by mesenchymal niche through VLA-4/CD44 axis. Leukemia 2014, 28, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef] [PubMed]
- Gado, K.; Domjan, G.; Hegyesi, H.; Falus, A. Role of INTERLEUKIN-6 in the pathogenesis of multiple myeloma. Cell Biol. Int. 2000, 24, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.F.; Lu, Z.Y.; Jourdan, M.; Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Munoz, J.; Garcia-Pardo, A.; Teixido, J. Molecular players in hematologic tumor cell trafficking. Front. Immunol. 2019, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Shimizu, N.; Dallas, M.; Niewolna, M.; Story, B.; Williams, P.J.; Mundy, G.R.; Yoneda, T. Anti-alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 2004, 104, 2149–2154. [Google Scholar] [CrossRef]
- Burger, R. Impact of interleukin-6 in hematological malignancies. Transfus. Med. Hemotherapy 2013, 40, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Portier, M.; Zhang, X.G.; Caron, E.; Lu, Z.Y.; Bataille, R.; Klein, B. gamma-Interferon in multiple myeloma: Inhibition of interleukin-6 (IL-6)-dependent myeloma cell growth and downregulation of IL-6-receptor expression in vitro. Blood 1993, 81, 3076–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Desai, P.; Shiao, R.T.; Lavelle, D.; Haleem, A.; Chen, J. Inhibition of myeloma cell growth by dexamethasone and all-trans retinoic acid: Synergy through modulation of interleukin-6 autocrine loop at multiple sites. Blood 1996, 87, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.; Chen, T.; Liu, J.; Dowling, P.; Hideshima, T.; Zhang, L.; Morelli, E.; Camci-Unal, G.; Wu, X.; Tai, Y.T.; et al. Targeting histone deacetylase 3 (HDAC3) in the bone marrow microenvironment inhibits multiple myeloma proliferation by modulating exosomes and IL-6 trans-signaling. Leukemia 2019. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; McMullan, C.; Poulaki, V.; Fanourakis, G.; Schlossman, R.; Chauhan, D.; Munshi, N.C.; Hideshima, T.M.; et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood 2003, 101, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Simmons, P.J.; Przepiorka, D.; Thomas, E.D.; Torok-Storb, B. Host origin of marrow stromal cells following allogeneic bone marrow transplantation. Nature 1987, 328, 429–432. [Google Scholar] [CrossRef]
- Marsh, J.C.; Harhalakis, N.; Dowding, C.; Laffan, M.; Gordon-Smith, E.C.; Hows, J.M. Recurrent graft failure following syngeneic bone marrow transplantation for aplastic anaemia. Bone Marrow Transplant. 1989, 4, 581–585. [Google Scholar]
- Witherspoon, R.P.; Schubach, W.; Neiman, P.; Martin, P.; Thomas, E.D. Donor cell leukemia developing six years after marrow grafting for acute leukemia. Blood 1985, 65, 1172–1174. [Google Scholar] [CrossRef]
- Lawler, M.; Locasciulli, A.; Longoni, D.; Schiro, R.; McCann, S.R. Leukaemic transformation of donor cells in a patient receiving a second allogeneic bone marrow transplant for severe aplastic anaemia. Bone Marrow Transplant. 2002, 29, 453–456. [Google Scholar] [CrossRef] [Green Version]
- McCann, S.R.; Bacigalupo, A.; Gluckman, E.; Hinterberger, W.; Hows, J.; Ljungman, P.; Marin, P.; Nissen, C.; van’t Veer Kerthof, E.; Raghavachar, A.; et al. Graft rejection and second bone marrow transplants for acquired aplastic anaemia: A report from the Aplastic Anaemia Working Party of the European Bone Marrow Transplant Group. Bone Marrow Transplant. 1994, 13, 233–237. [Google Scholar]
- Cavazzana, M.; Bushman, F.D.; Miccio, A.; Andre-Schmutz, I.; Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: Progress and challenges. Nat. Rev. Drug Discov. 2019, 18, 447–462. [Google Scholar] [CrossRef] [Green Version]
- Cavazzana, M.; Ribeil, J.A.; Lagresle-Peyrou, C.; Andre-Schmutz, I. Gene therapy with hematopoietic stem cells: The diseased bone marrow’s point of view. Stem Cells Dev. 2017, 26, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, N.A.; Malani, N.; Melamed, A.; Gormley, N.; Carter, R.; Bentley, D.; Berry, C.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood 2011, 117, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.H.; Del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; García-García, A.; Macías, D.; González-Gómez, C.; Del Monte, A.; Wittner, M.; et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell 2019, 25, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiuti, A.; Naldini, L. Safer conditioning for blood stem cell transplants. Nat. Biotechnol. 2016, 34, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Ingo, D.M.; Redaelli, D.; Rossella, V.; Perini, O.; Santoleri, L.; Ciceri, F.; Aiuti, A.; Bernardo, M.E. Bone marrow-derived CD34(-) fraction: A rich source of mesenchymal stromal cells for clinical application. Cytotherapy 2016, 18, 1560–1563. [Google Scholar] [CrossRef] [PubMed]
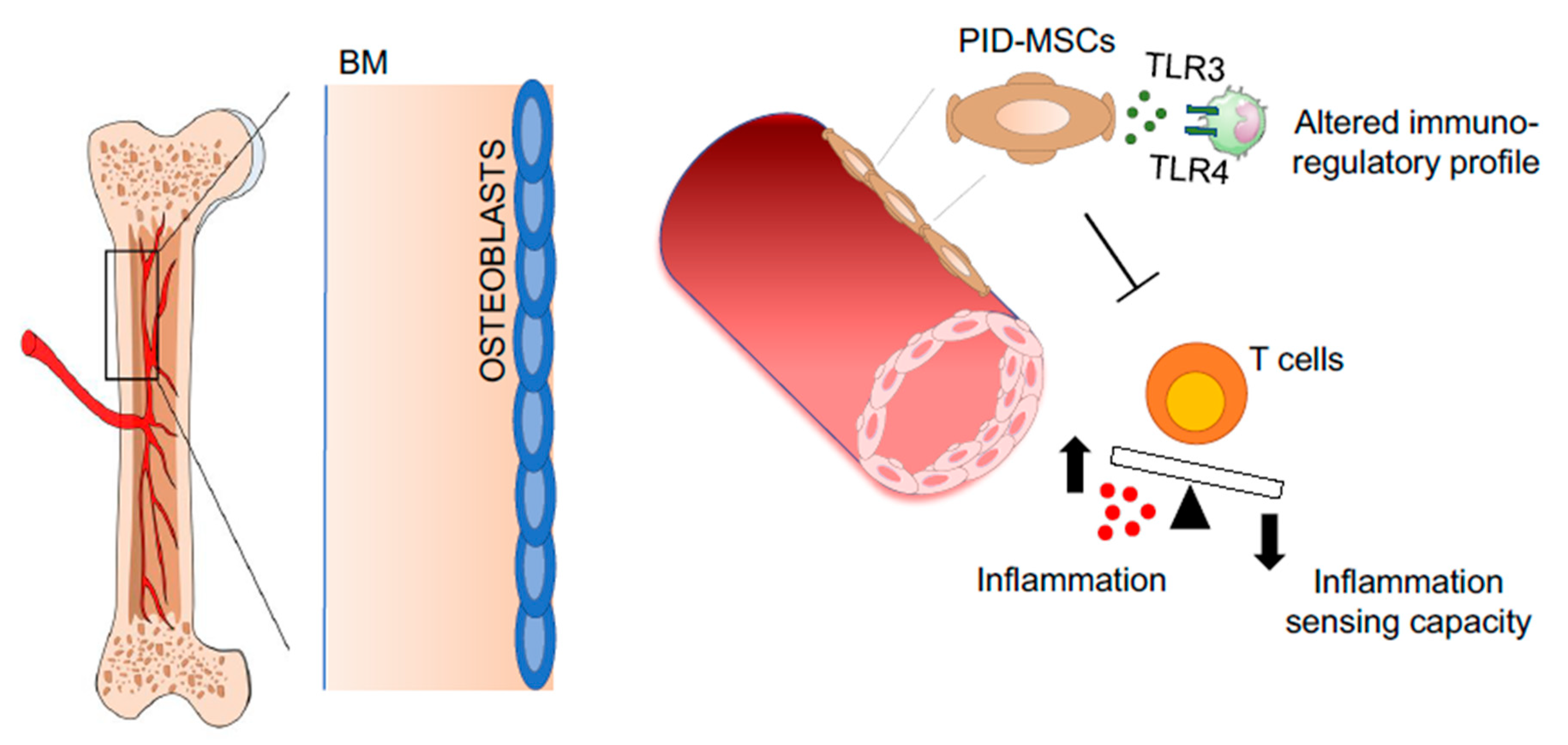
- Starc, N.; Ingo, D.; Conforti, A.; Rossella, V.; Tomao, L.; Pitisci, A.; De Mattia, F.; Brigida, I.; Algeri, M.; Montanari, M.; et al. Biological and functional characterization of bone marrow-derived mesenchymal stromal cells from patients affected by primary immunodeficiency. Sci. Rep. 2017, 7, 8153. [Google Scholar] [CrossRef] [Green Version]
- Weisser, M.; Demel, U.M.; Stein, S.; Chen-Wichmann, L.; Touzot, F.; Santilli, G.; Sujer, S.; Brendel, C.; Siler, U.; Cavazzana, M.; et al. Hyperinflammation in patients with chronic granulomatous disease leads to impairment of hematopoietic stem cell functions. J. Allergy Clin. Immunol. 2016, 138, 219–228. [Google Scholar] [CrossRef] [Green Version]
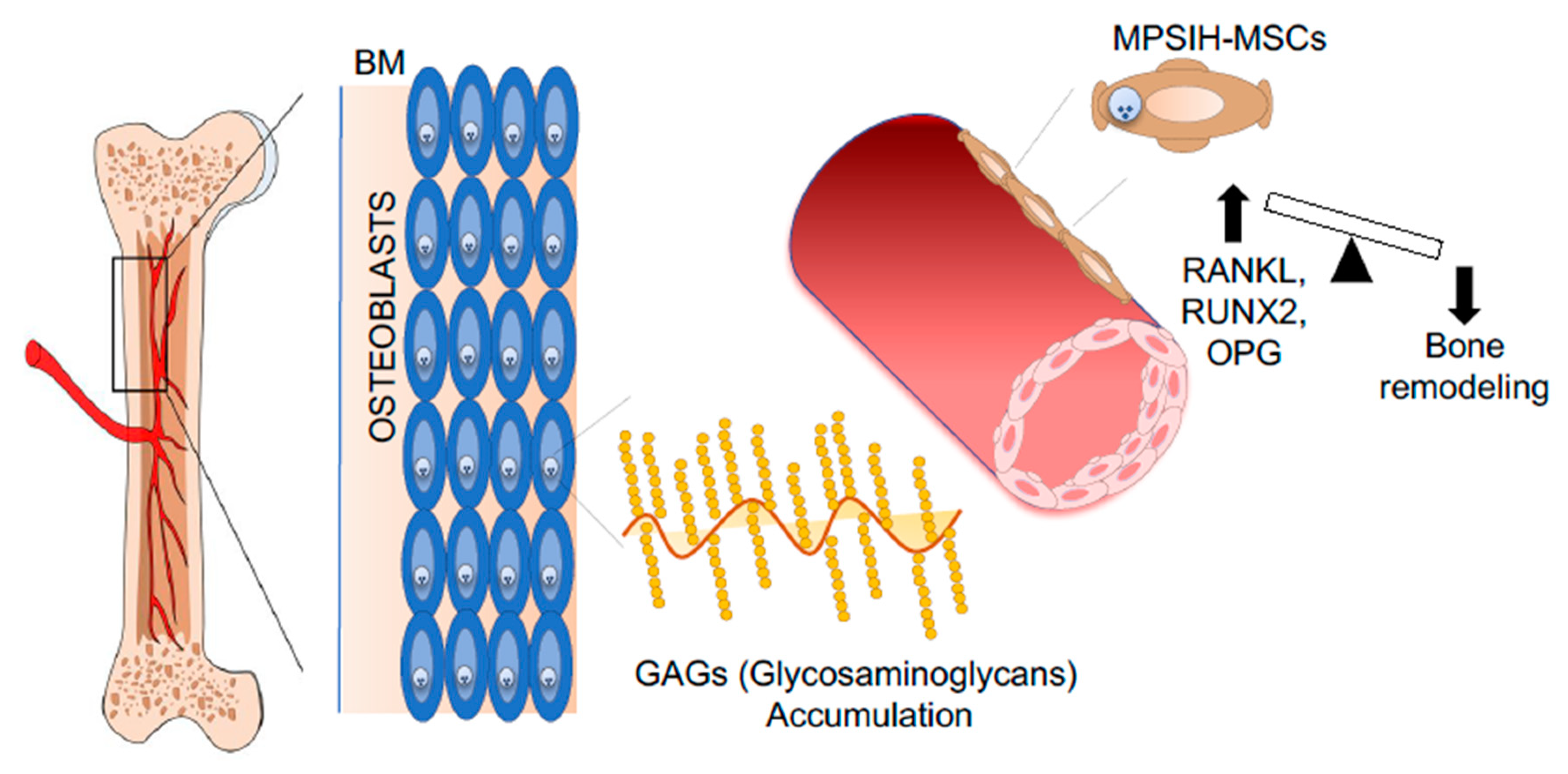
- Giugliani, R.; Federhen, A.; Rojas, M.V.; Vieira, T.; Artigalás, O.; Pinto, L.L.; Azevedo, A.C.; Acosta, A.; Bonfim, C.; Lourenço, C.M.; et al. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genet. Mol. Biol. 2010, 33, 589–604. [Google Scholar] [CrossRef]
- Gatto, F.; Redaelli, D.; Salvadè, A.; Marzorati, S.; Sacchetti, B.; Ferina, C.; Roobrouck, V.D.; Bertola, F.; Romano, M.; Villani, G.; et al. Hurler disease bone marrow stromal cells exhibit altered ability to support osteoclast formation. Stem Cells Dev. 2012, 21, 1466–1477. [Google Scholar] [CrossRef] [Green Version]
- Takamatsu, Y.; Simmons, P.J.; Moore, R.J.; Morris, H.A.; To, L.B.; Lévesque, J.P. Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood 1998, 92, 3465–3473. [Google Scholar] [CrossRef] [PubMed]
- Lymperi, S.; Ersek, A.; Ferraro, F.; Dazzi, F.; Horwood, N.J. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood 2011, 117, 1540–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, A.; Abou-Ezzi, G.; Sitnicka, E.; Jacobsen, S.E.; Wakkach, A.; Blin-Wakkach, C. Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J. Exp. Med. 2012, 209, 537–549. [Google Scholar] [CrossRef] [PubMed]
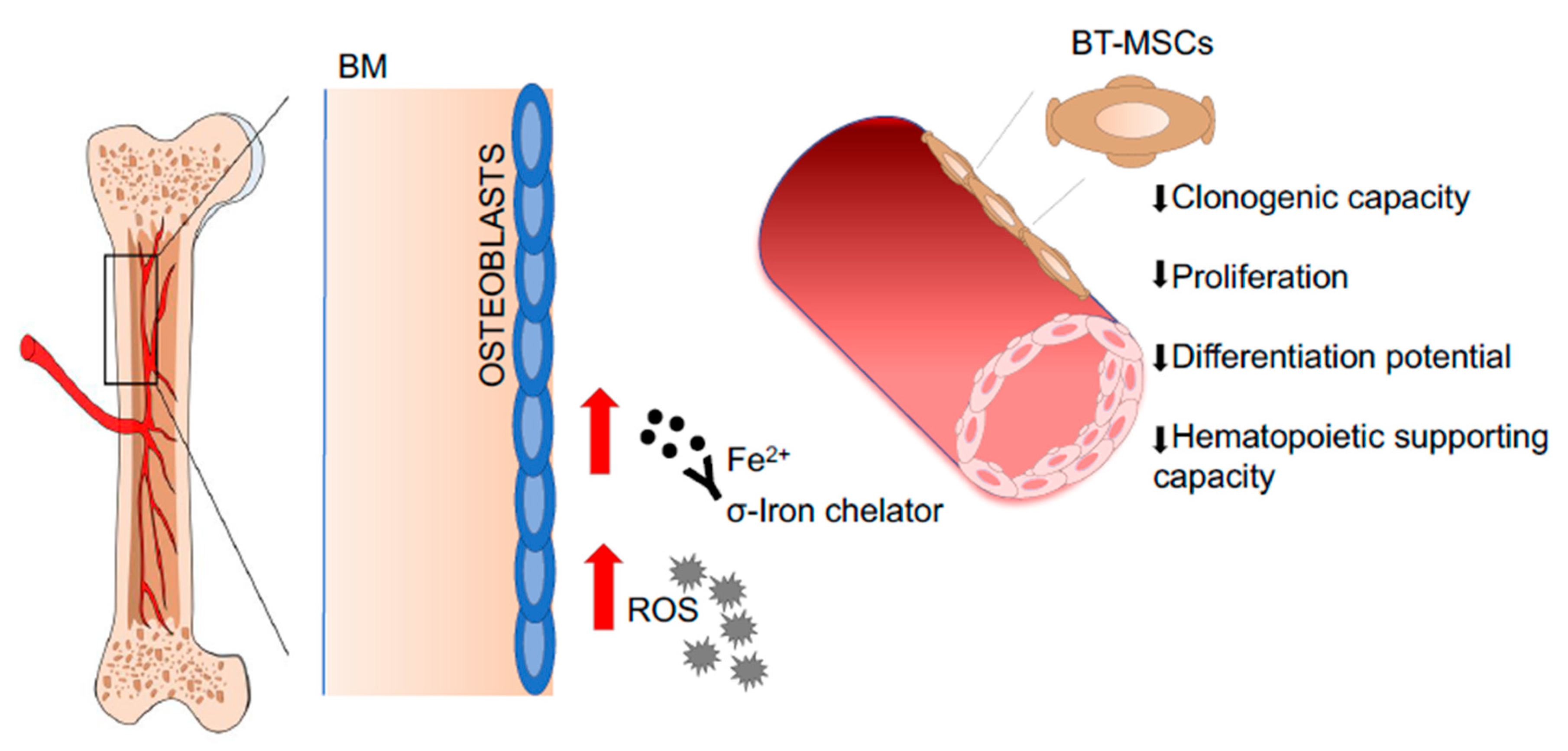
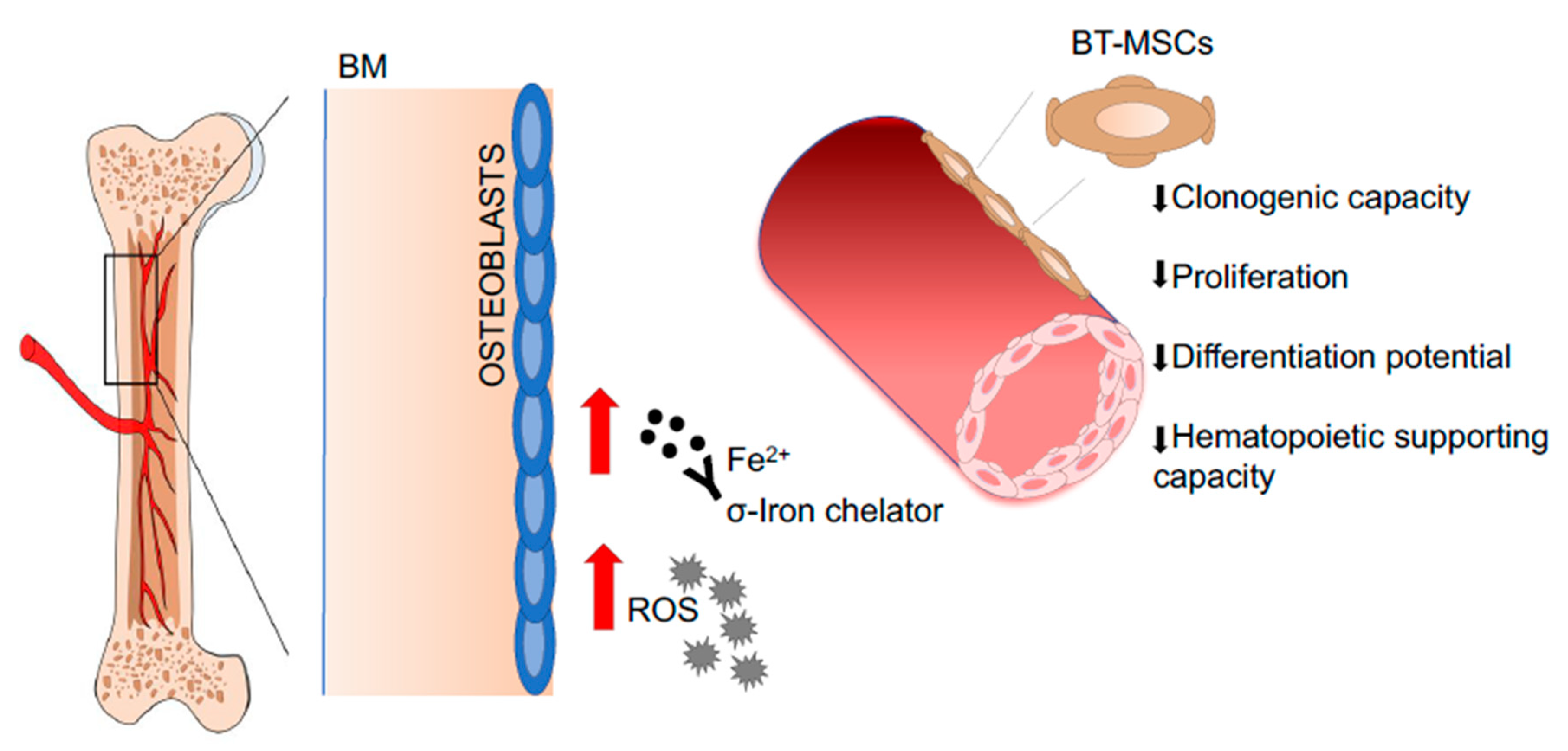
- Crippa, S.; Rossella, V.; Aprile, A.; Silvestri, L.; Rivis, S.; Scaramuzza, S.; Pirroni, S.; Avanzini, M.A.; Basso-Ricci, L.; Hernandez, R.J.; et al. Bone marrow stromal cells from beta-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Investig. 2019, 129, 1566–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaziev, D.; Polchi, P.; Galimberti, M.; Angelucci, E.; Giardini, C.; Baronciani, D.; Erer, B.; Lucarelli, G. Graft-versus-host disease after bone marrow transplantation for thalassemia: An analysis of incidence and risk factors. Transplantation 1997, 63, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Fouzia, N.; Edison, E.; Lakshmi, K.M.; Korula, A.; Velayudhan, S.R.; Balasubramanian, P.; Abraham, A.; Viswabandya, A.; George, B.; Mathews, V.; et al. Long-term outcome of mixed chimerism after stem cell transplantation for thalassemia major conditioned with busulfan and cyclophosphamide. Bone Marrow Transplant. 2018, 53, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Xiao, H.; Yao, Z.; Zhang, H.; Liu, Q.; Wu, B.; Nie, D.; Li, Y.; Pang, Y.; et al. Cotransplantation of bone marrow-derived mesenchymal stem cells in haploidentical hematopoietic stem cell transplantation in patients with severe aplastic anemia: An interim summary for a multicenter phase II trial results. Bone Marrow Transplant. 2017, 52, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagesan, S.; Griffin, M.D. Autologous and allogeneic mesenchymal stem cells in organ transplantation: What do we know about their safety and efficacy? Curr. Opin. Organ. Transplant. 2014, 19, 65–72. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crippa, S.; Santi, L.; Bosotti, R.; Porro, G.; Bernardo, M.E. Bone Marrow-Derived Mesenchymal Stromal Cells: A Novel Target to Optimize Hematopoietic Stem Cell Transplantation Protocols in Hematological Malignancies and Rare Genetic Disorders. J. Clin. Med. 2020, 9, 2. https://doi.org/10.3390/jcm9010002
Crippa S, Santi L, Bosotti R, Porro G, Bernardo ME. Bone Marrow-Derived Mesenchymal Stromal Cells: A Novel Target to Optimize Hematopoietic Stem Cell Transplantation Protocols in Hematological Malignancies and Rare Genetic Disorders. Journal of Clinical Medicine. 2020; 9(1):2. https://doi.org/10.3390/jcm9010002
Chicago/Turabian StyleCrippa, Stefania, Ludovica Santi, Roberto Bosotti, Giulia Porro, and Maria Ester Bernardo. 2020. "Bone Marrow-Derived Mesenchymal Stromal Cells: A Novel Target to Optimize Hematopoietic Stem Cell Transplantation Protocols in Hematological Malignancies and Rare Genetic Disorders" Journal of Clinical Medicine 9, no. 1: 2. https://doi.org/10.3390/jcm9010002