Highly Sensitive Detection of IDH2 Mutations in Acute Myeloid Leukemia

 ,
,  ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Patient’s Cohort
2.2. Cloning PCR Controls with pGEM®—T Easy Vector
2.3. Sanger Sequencing for IDH2mut Detection
2.4. ddPCR for IDH2mut Detection
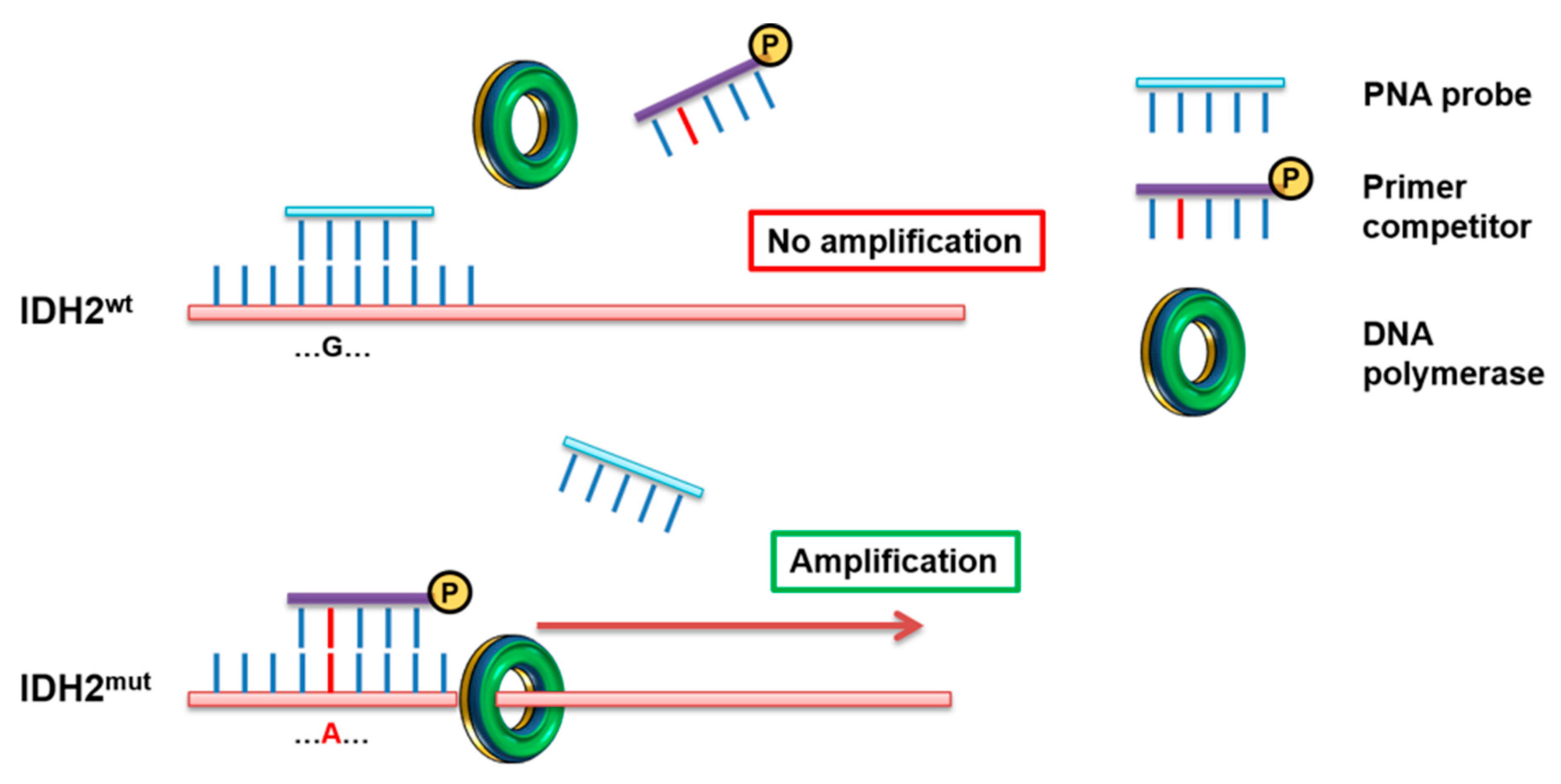
2.5. PNA Directed PCR Clamping for IDH2mut Detection
2.6. Statistical Analysis
3. Results
3.1. IDH2 R140Q and R172K Detection by PNA-PCR Clamping
3.2. Comparison of Sanger Sequencing, PNA-PCR Clamping and ddPCR for the IDH2mut Detection
3.3. Prevalence of IDH2mut in AML Samples
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Song, X.; Peng, Y.; Wang, X.; Chen, Y.; Jin, L.; Yang, T.; Qian, M.; Ni, W.; Tong, X.; Lan, J. Incidence, Survival, and Risk Factors for Adults with Acute Myeloid Leukemia Not Otherwise Specified and Acute Myeloid Leukemia with Recurrent Genetic Abnormalities: Analysis of the Surveillance, Epidemiology, and End Results (SEER) Database, 2001–2013. Acta Haematol. 2018, 139, 115–127. [Google Scholar] [CrossRef]
- Deschler, B.; Lubbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef]
- Juliusson, G.; Lazarevic, V.; Horstedt, A.S.; Hagberg, O.; Hoglund, M.; Swedish Acute Leukemia Registry Group. Acute myeloid leukemia in the real world: Why population-based registries are needed. Blood 2012, 119, 3890–3899. [Google Scholar] [CrossRef] [Green Version]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Su, R.; Weng, H.; Huang, H.; Li, Z.; Chen, J. RNA N(6)-methyladenosine modification in cancers: Current status and perspectives. Cell Res. 2018, 28, 507–517. [Google Scholar] [CrossRef]
- Debarri, H.; Lebon, D.; Roumier, C.; Cheok, M.; Marceau-Renaut, A.; Nibourel, O.; Geffroy, S.; Helevaut, N.; Rousselot, P.; Gruson, B.; et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: A study by the Acute Leukemia French Association. Oncotarget 2015, 6, 42345–42353. [Google Scholar] [CrossRef] [Green Version]
- Ok, C.Y.; Loghavi, S.; Sui, D.; Wei, P.; Kanagal-Shamanna, R.; Yin, C.C.; Zuo, Z.; Routbort, M.J.; Tang, G.; Tang, Z.; et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica 2019, 104, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Cilloni, D.; Petiti, J.; Rosso, V.; Andreani, G.; Dragani, M.; Fava, C.; Saglio, G. Digital PCR in Myeloid Malignancies: Ready to Replace Quantitative PCR? Int. J. Mol. Sci. 2019, 20, 2249. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Kyger, E.M.; Krevolin, M.D.; Powell, M.J. Detection of the hereditary hemochromatosis gene mutation by real-time fluorescence polymerase chain reaction and peptide nucleic acid clamping. Anal. Biochem. 1998, 260, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Petiti, J.; Rosso, V.; Lo Iacono, M.; Calabrese, C.; Signorino, E.; Gaidano, V.; Berger, M.; Saglio, G.; Cilloni, D. Prognostic significance of The Wilms’ Tumor-1 (WT1) rs16754 polymorphism in acute myeloid leukemia. Leuk. Res. 2018, 67, 6–11. [Google Scholar] [CrossRef]
- Rosso, V.; Bracco, E.; Pedrola, R.; Carturan, S.; Signorino, E.; Petiti, J.; Calabrese, C.; Nicoli, P.; De Gobbi, M.; Gaidano, V.; et al. Detection of BCR-ABL T315I mutation by peptide nucleic acid directed PCR clamping and by peptide nucleic acid FISH. Biomark. Res. 2015, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Rosso, V.; Petiti, J.; Bracco, E.; Pedrola, R.; Carnuccio, F.; Signorino, E.; Carturan, S.; Calabrese, C.; Bot-Sartor, G.; Ronconi, M.; et al. A novel assay to detect calreticulin mutations in myeloproliferative neoplasms. Oncotarget 2017, 8, 6399–6405. [Google Scholar] [CrossRef] [Green Version]
- Gorello, P.; Cazzaniga, G.; Alberti, F.; Dell’Oro, M.G.; Gottardi, E.; Specchia, G.; Roti, G.; Rosati, R.; Martelli, M.F.; Diverio, D.; et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 2006, 20, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Gabert, J.; Beillard, E.; van der Velden, V.H.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cave, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Hasan, S.K.; Ottone, T.; Lavorgna, S.; Ciardi, C.; Angelini, D.F.; Agostini, F.; Venditti, A.; Lo-Coco, F. Two novel methods for rapid detection and quantification of DNMT3A R882 mutations in acute myeloid leukemia. J. Mol. Diagn. 2015, 17, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: A European LeukemiaNet study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [Green Version]
- Oliner, K.; Juan, T.; Suggs, S.; Wolf, M.; Sarosi, I.; Freeman, D.J.; Gyuris, T.; Baron, W.; Bakker, A.; Parker, A.; et al. A comparability study of 5 commercial KRAS tests. Diagn. Pathol. 2010, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.P.; Ravandi, F.; Ma, D.; Paladugu, A.; Barkoh, B.A.; Medeiros, L.J.; Luthra, R. Acute myeloid leukemia with IDH1 or IDH2 mutation: Frequency and clinicopathologic features. Am. J. Clin. Pathol. 2011, 135, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Chotirat, S.; Thongnoppakhun, W.; Promsuwicha, O.; Boonthimat, C.; Auewarakul, C.U. Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J. Hematol. Oncol. 2012, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Petrova, L.; Vrbacky, F.; Lanska, M.; Zavrelova, A.; Zak, P.; Hrochova, K. IDH1 and IDH2 mutations in patients with acute myeloid leukemia: Suitable targets for minimal residual disease monitoring? Clin. Biochem. 2018, 61, 34–39. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer/PNA Probe | Sequence 5′-3′ |
|---|---|
| IDH2 forward | CCAATGGAACTATCCAGAACATC |
| IDH2 R140Q reverse | CTCGTCGGTGTTGTACATGC |
| IDH2 R172K reverse | TATATCGCCATGGGCGTGCTT |
| IDH2 R140Q PNA | CTATCCGGAACATCCT |
| IDH2 R172K PNA | TGGGCGTGCCTGCCAAT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petiti, J.; Rosso, V.; Croce, E.; Franceschi, V.; Andreani, G.; Dragani, M.; De Gobbi, M.; Lunghi, M.; Saglio, G.; Fava, C.; et al. Highly Sensitive Detection of IDH2 Mutations in Acute Myeloid Leukemia. J. Clin. Med. 2020, 9, 271. https://doi.org/10.3390/jcm9010271
Petiti J, Rosso V, Croce E, Franceschi V, Andreani G, Dragani M, De Gobbi M, Lunghi M, Saglio G, Fava C, et al. Highly Sensitive Detection of IDH2 Mutations in Acute Myeloid Leukemia. Journal of Clinical Medicine. 2020; 9(1):271. https://doi.org/10.3390/jcm9010271
Chicago/Turabian StylePetiti, Jessica, Valentina Rosso, Eleonora Croce, Vanessa Franceschi, Giacomo Andreani, Matteo Dragani, Marco De Gobbi, Monia Lunghi, Giuseppe Saglio, Carmen Fava, and et al. 2020. "Highly Sensitive Detection of IDH2 Mutations in Acute Myeloid Leukemia" Journal of Clinical Medicine 9, no. 1: 271. https://doi.org/10.3390/jcm9010271
APA StylePetiti, J., Rosso, V., Croce, E., Franceschi, V., Andreani, G., Dragani, M., De Gobbi, M., Lunghi, M., Saglio, G., Fava, C., Lo Iacono, M., & Cilloni, D. (2020). Highly Sensitive Detection of IDH2 Mutations in Acute Myeloid Leukemia. Journal of Clinical Medicine, 9(1), 271. https://doi.org/10.3390/jcm9010271