Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders

 ,
,
Abstract
:1. Introduction
2. Materials & Methods
3. Results
3.1. Clinical Presentation of the Four Patients
3.1.1. Patient 1. Myasthenic Syndrome, Congenital, 22 (OMIM 616224)
3.1.2. Patient 2. Autosomal Recessive Dopa-Responsive Dystonia (OMIM 605407)
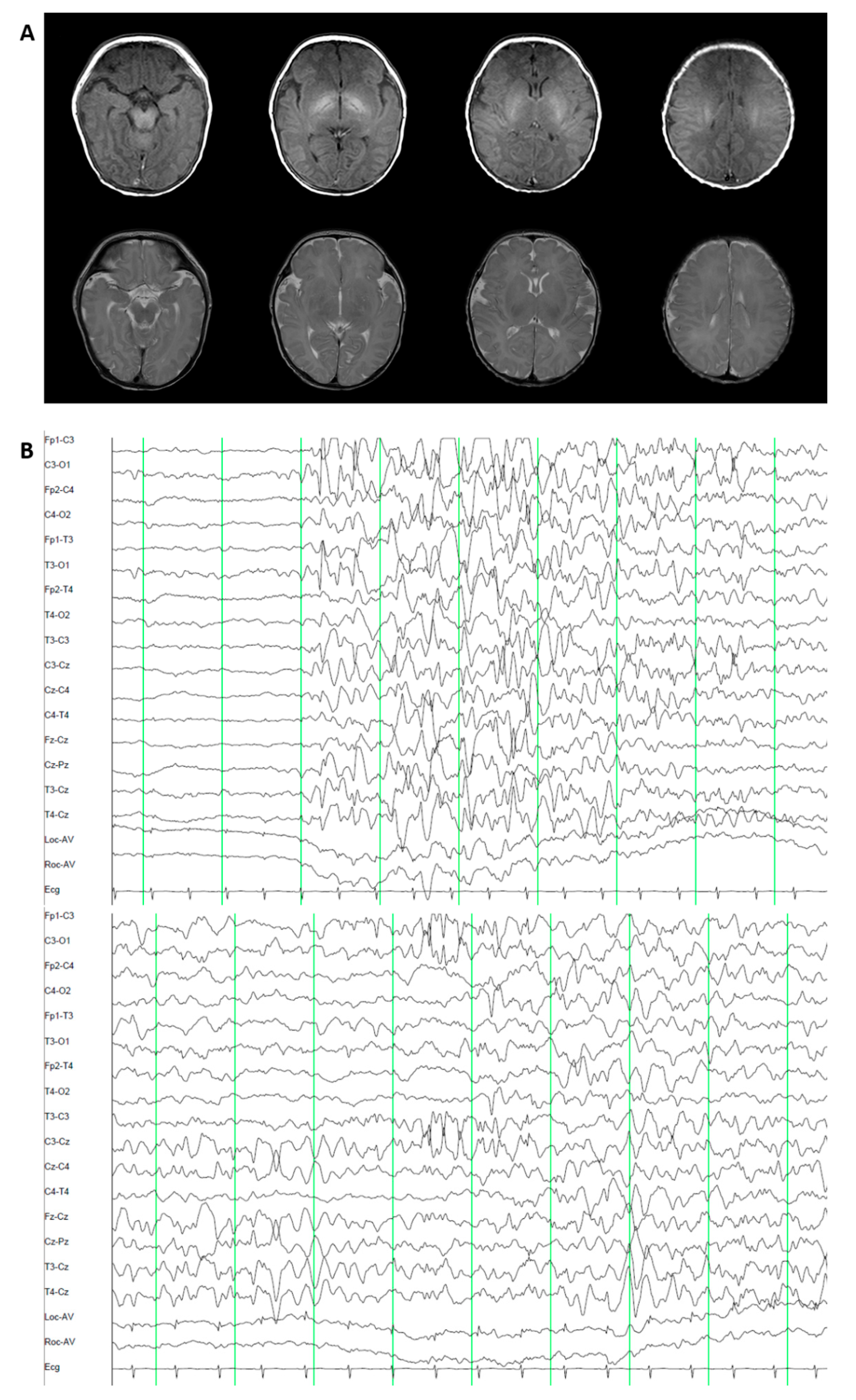
3.1.3. Patient 3. Epileptic Encephalopathy 47 (OMIM 617166)
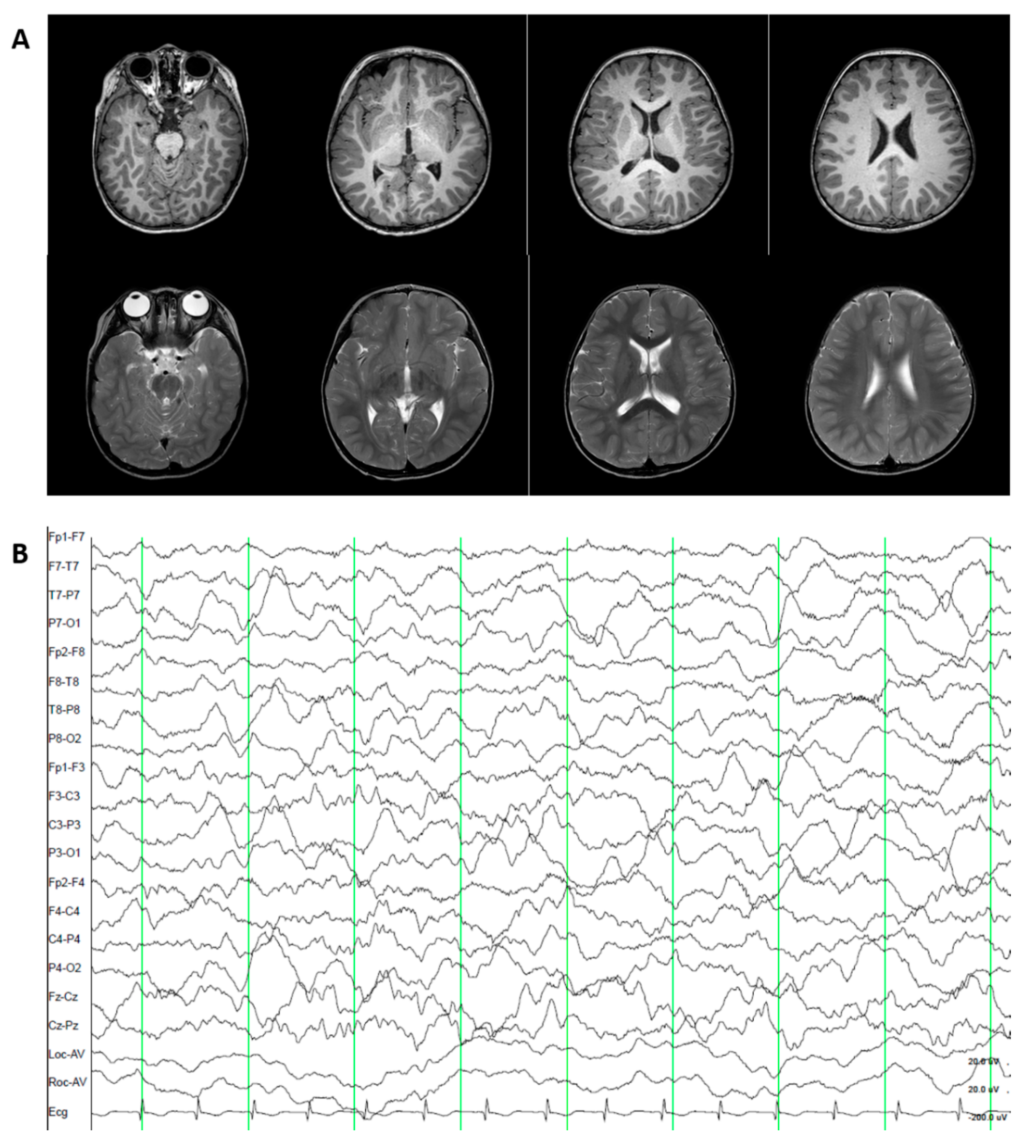
3.1.4. Patient 4. Episodic Ataxia, Type II (OMIM 108500).
3.2. Genetic Diagnosis
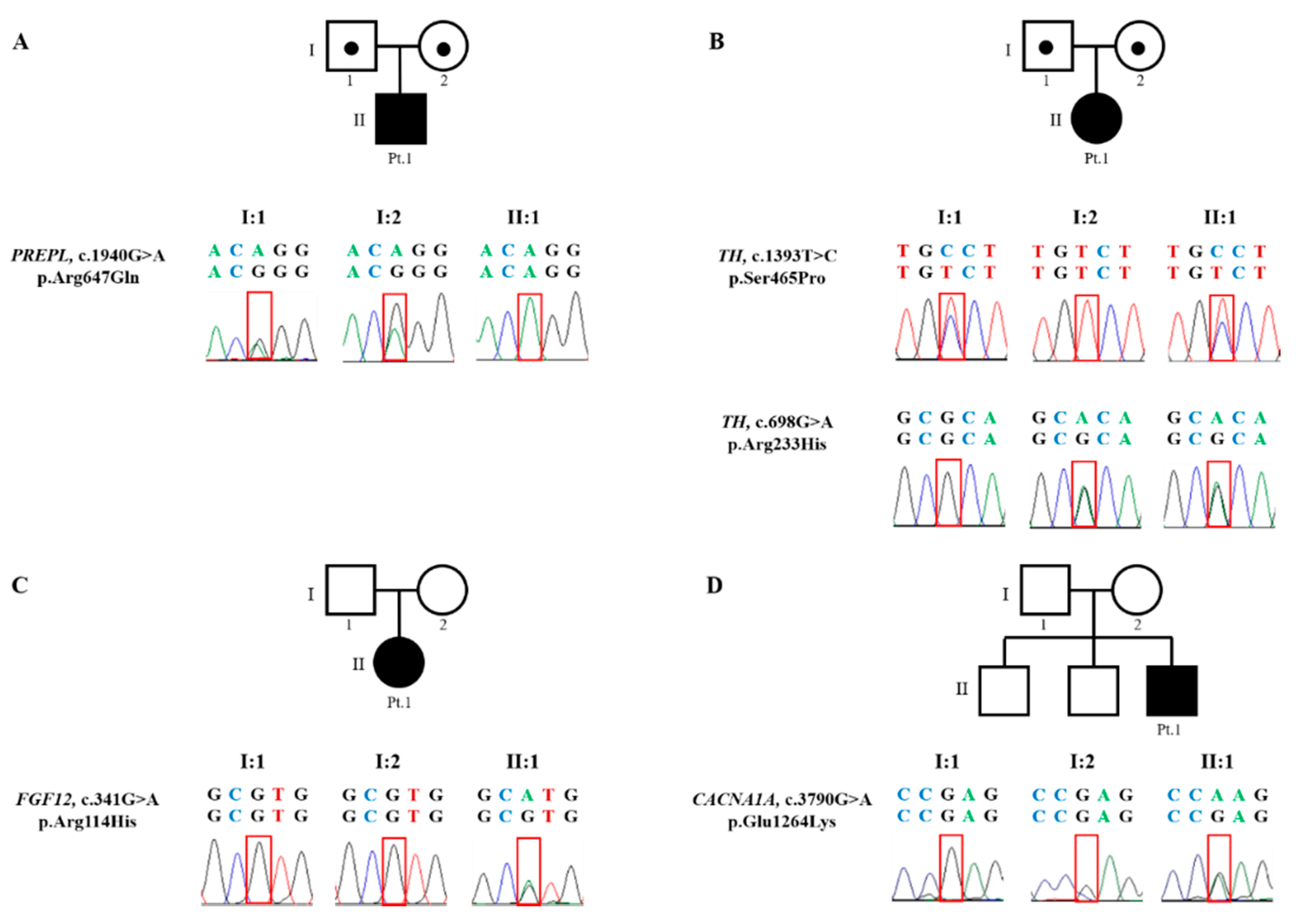
3.2.1. Patient 1. Myasthenic Syndrome, Congenital, 22 (OMIM 616224)
3.2.2. Patient 2. Autosomal Recessive Dopa-Responsive Dystonia (OMIM 605407)
3.2.3. Patient 3. Epileptic Encephalopathy 47 (OMIM 617166)
3.2.4. Patient 4. Episodic Ataxia, Type II (OMIM 108500)
3.3. Post-Diagnosis Treatment and Clinical Outcome
3.3.1. Patient 1. Myasthenic Syndrome, Congenital, 22 (OMIM 616224)
3.3.2. Patient 2. Autosomal Recessive Dopa-Responsive Dystonia (OMIM 605407)
3.3.3. Patient 3. Epileptic Encephalopathy 47 (OMIM 617166)
3.3.4. Patient 4. Episodic Ataxia, Type II (OMIM 108500)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.F.; Fitzpatrick, D.R.; Firth, H.V. Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 2018, 19, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Zhou, L.; Zheng, M.-Y.; Huang, J.; Wan, S.; Zhu, A.; Zhang, M.; Dong, A.; Hou, L.; Li, J.; et al. Diagnostic and clinical utility of whole genome sequencing in a cohort of undiagnosed Chinese families with rare diseases. Sci. Rep. 2019, 9, 19365. [Google Scholar] [CrossRef]
- Posey, J.E. Genome sequencing and implications for rare disorders. Orphanet J. Rare Dis. 2019, 14, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaLonde, E.; Rentas, S.; Lin, F.; Dulik, M.C.; Skraban, C.M.; Spinner, N.B. Genomic Diagnosis for Pediatric Disorders: Revolution and Evolution. Front. Pediatr. 2020, 8, 373. [Google Scholar] [CrossRef]
- Haskell, G.T.; Adams, M.C.; Fan, Z.; Amin, K.; Badillo, R.J.G.; Zhou, L.; Bizon, C.; Chahin, N.; Greenwood, R.S.; Milko, L.V.; et al. Diagnostic utility of exome sequencing in the evaluation of neuromuscular disorders. Neurol. Genet. 2018, 4, e212. [Google Scholar] [CrossRef] [Green Version]
- Rexach, J.; Lee, H.; Martinez-Agosto, J.; Németh, A.H.; Fogel, B.L. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019, 18, 492–503. [Google Scholar] [CrossRef]
- Federico, A. Rare neurological diseases: A Pandora’s box for neurology (an European and Italian perspective). Rev. Neurol. 2013, 169, S12–S17. [Google Scholar] [CrossRef]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Seo, G.H.; Kim, T.; Choi, I.H.; Park, J.-Y.; Lee, J.; Kim, S.; Won, D.-G.; Oh, A.; Lee, Y.; Choi, J.; et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, L.P.; Luiten, B.; Smeitink, J.A.M.; Andel, J.F.D.R.-V.; Hyland, K.; Steenbergen-Spanjers, G.C.H.; Janssen, R.J.T.; Wevers, R.A. A common point mutation in the tyrosine hydroxylase gene in autosomal recessive L-DOPA-responsive dystonia in the Dutch population. Hum. Genet. 1998, 102, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, M. Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell. Mol. Life Sci. 2012, 69, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siekierska, A.; Isrie, M.; Liu, Y.; Scheldeman, C.; Vanthillo, N.; Lagae, L.; De Witte, P.A.; Van Esch, H.; Goldfarb, M.; Buyse, G.M. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology 2016, 86, 2162–2170. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Al-Mehmadi, S.; Splitt, M.; Ramesh, V.; Debrosse, S.; Dessoffy, K.; Xia, F.; Yang, Y.; Rosenfeld, J.A.; Cossette, P.; Michaud, J.L.; et al. FHF1 (FGF12) epileptic encephalopathy. Neurol. Genet. 2016, 2, e115. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995, 64, 493–531. [Google Scholar] [CrossRef]
- Llinas, R.; Sugimori, M.; Hillman, D.; Cherksey, B. Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous system. Trends Neurosci. 1992, 15, 351–355. [Google Scholar] [CrossRef]
- Ginsburg, G.S.; Phillips, K.A. Precision Medicine: From Science To Value. Health Aff. 2018, 37, 694–701. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Shyr, C.; Ross, C.J.; Horvath, G.A.; Salvarinova, R.; Ye, X.C.; Zhang, L.-H.; Bhavsar, A.P.; Lee, J.J.; Drögemöller, B.I.; et al. Exome Sequencing and the Management of Neurometabolic Disorders. N. Engl. J. Med. 2016, 374, 2246–2255. [Google Scholar] [CrossRef] [Green Version]
- Demos, M.; Guella, I.; DeGuzman, C.; McKenzie, M.B.; Buerki, S.E.; Evans, D.M.; Toyota, E.B.; Boelman, C.; Huh, L.L.; Datta, A.; et al. Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy. Front. Neurol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Tambuyzer, E.; VandenDriessche, B.; Austin, C.P.; Brooks, P.J.; Larsson, K.; Needleman, K.I.M.; Valentine, J.; Davies, K.E.; Groft, S.C.; Preti, R.; et al. Therapies for rare diseases: Therapeutic modalities, progress and challenges ahead. Nat. Rev. Drug Discov. 2020, 19, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Bruggemann, N.; Klein, C. Will genotype drive treatment options? Mov. Disord. 2019, 34, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Cohen, J.S.; Vernon, H.; Barañano, K.; McClellan, R.; Jamal, L.; Naidu, S.; Fatemi, A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014, 76, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.-M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Bestue-Cardiel, M.; Natera-de Benito, D. Current status of congenital myasthenic syndromes. Rev. Neurol. 2017, 65, 161–176. (In Spanish) [Google Scholar] [PubMed]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Lone, A.M.; Leidl, M.; McFedries, A.K.; Horner, J.W.; Creemers, J.; Saghatelian, A. Deletion of Prepl Causes Growth Impairment and Hypotonia in Mice. PLoS ONE 2014, 9, e89160. [Google Scholar] [CrossRef] [Green Version]
- Laugwitz, L.; Redler, S.; Buchert, R.; Sturm, M.; Zeile, I.; Schara, U.; Wieczorek, D.; Haack, T.; Distelmaier, F. Isolated PREPL deficiency associated with congenital myasthenic syndrome-22. Klin. Padiatr. 2018, 230, 281–283. [Google Scholar] [CrossRef]
- Regal, L.; Aydin, H.I.; Dieltjens, A.-M.; Van Esch, H.; Francois, I.; Okur, I.; Zeybek, C.; Meulemans, S.; Van Mol, C.; Van Bruwaene, L.; et al. Two novel deletions in hypotonia-cystinuria syndrome. Mol. Genet. Metab. 2012, 107, 614–616. [Google Scholar] [CrossRef]
- Yang, Q.; Hua, R.; Qian, J.; Yi, S.; Shen, F.; Zhang, Q.; Li, M.; Yi, S.; Luo, J.; Fan, X. PREPL Deficiency: A Homozygous Splice Site PREPL Mutation in a Patient With Congenital Myasthenic Syndrome and Absence of Ovaries and Hypoplasia of Uterus. Front. Genet. 2020, 11, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regal, L.; Martensson, E.; Maystadt, I.; Voermans, N.; Lederer, D.; Burlina, A.; Fita, M.J.J.; Hoogeboom, A.J.M.; Engman, M.O.; Hollemans, T.; et al. PREPL deficiency: Delineation of the phenotype and development of a functional blood assay. Genet. Med. 2018, 20, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regal, L.; Shen, X.-M.; Selcen, D.; Verhille, C.; Meulemans, S.; Creemers, J.W.M.; Engel, A.G. PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology 2014, 82, 1254–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, S.; Miyake, N.; Tapia, C.; Matsumoto, N. The second point mutation in PREPL: A case report and literature review. J. Hum. Genet. 2018, 63, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Ludecke, B.; Dworniczak, B.; Bartholome, K. A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum. Genet. 1995, 95, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.-J.; Andel, J.F.D.R.-V.; Aeby, A.; Blau, N.; Burlina, A.; Donati, M.A.; Geurtz, B.; Grattan-Smith, P.J.; et al. Tyrosine hydroxylase deficiency: A treatable disorder of brain catecholamine biosynthesis. Brain 2010, 133 Pt 6, 1810–1822. [Google Scholar] [CrossRef] [Green Version]
- Yeung, W.L.; Wong, V.C.N.; Chan, K.-Y.; Hui, J.; Fung, C.-W.; Yau, E.; Ko, C.-H.; Lam, C.-W.; Mak, C.M.; Siu, S.; et al. Expanding Phenotype and Clinical Analysis of Tyrosine Hydroxylase Deficiency. J. Child Neurol. 2011, 26, 179–187. [Google Scholar] [CrossRef]
- Katus, L.E.; Frucht, S.J. An unusual presentation of tyrosine hydroxylase deficiency. J. Clin. Mov. Disord. 2017, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.E.; Bally, J.F.; Breen, D.P.; Schaake, S.; Trinh, J.; Rakovic, A.; Klein, C. Mild dopa-responsive dystonia in heterozygous tyrosine hydroxylase mutation carrier: Evidence of symptomatic enzyme deficiency? Response from the authors. Parkinsonism Relat. Disord. 2020, 74, 80. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Meletti, S.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Noh, G.J.; Asher, Y.J.T.; Graham, J.M., Jr. Clinical review of genetic epileptic encephalopathies. Eur. J. Med Genet. 2012, 55, 281–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, D.K.; Pong, A.W.; Chung, W.K. Genetic evaluation and counseling for epilepsy. Nat. Rev. Neurol. 2010, 6, 445–453. [Google Scholar] [CrossRef] [PubMed]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Jen, J.; Graves, T.; Hess, E.; Hanna, M.; Griggs, R.; Baloh, R. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain 2007, 130 Pt 10, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Garone, G.; Capuano, A.; Travaglini, L.; Graziola, F.; Stregapede, F.; Zanni, G.; Vigevano, F.; Bertini, E.; Nicita, F. Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. Int. J. Mol. Sci. 2020, 21, 3603. [Google Scholar] [CrossRef] [PubMed]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; Van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.; Mohrenweiser, H.W.; Bulman, D.; Ferrari, M.; et al. Familial Hemiplegic Migraine and Episodic Ataxia Type-2 Are Caused by Mutations in the Ca2+ Channel Gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Kotagal, V. Acetazolamide-Responsive Ataxia. Semin. Neurol. 2012, 32, 533–537. [Google Scholar] [CrossRef]
- Hess, E.; Strupp, M.; Claassen, J.; Kalla, R.; Jen, J. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011, 77, 1996–1997. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Sex | Male | Female | Female | Male |
| Main neurologic sign and symptom | Hypotonia | Global delayed development, hypotonia | Seizure, developmental and epileptic encephalopathy | Episodic ataxia, Global delayed development |
| Symptom onset age | Perinatal period | Infant period | Perinatal period | Infant period |
| Final diagnosis | Congenital myasthenic syndrome 22 | Dopa-responsive dystonia | Epileptic encephalopathy 47 | Episodic ataxia type II |
| Age at diagnosis, year | 2.6 | 0.8 | 8.0 | 4.0 |
| Family history | None | None | None | None |
| Multisystemic involvement | Dysmorphic face, hearing disturbance, aortic stenosis, cryptorchidism, duplex kidney | None | None | None |
| Involved gene | Homozygous variant PREPL gene (NM_001171603.1) p.Arg647Gln | Heterozygous variants TH gene (NM_199292.2) p.Ser465Pro from father p.Arg233His from mother | Heterozygous variants de novo FGF12 gene (NM_021032.4) p.Arg114His | Heterozygous variants de novo CACNA1A gene (NM_001127221.1) p.Glu1264Lys |
| gnomAD (total population frequency) | 0.00008501 | 0.000003992 and 0.0001160 | absent | absent |
| In silico score | MutationTaster; Deleterious (1) Polyphen-2:Probably damaging (1) SIFT: Damaging (0) | REVEL: 0.846/0.955 | REVEL: 0.548 | REVEL: 0.922 |
| Chromosomal location | 2q21 | 11p15.5 | 3q28-q29 | 19p13.13 |
| Inheritance pattern | AR | AR | AD | AD |
| Treatment | Pyridostigmine | Levodopa | Phenytoin | Acetazolamide |
| Patients | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Average |
|---|---|---|---|---|---|
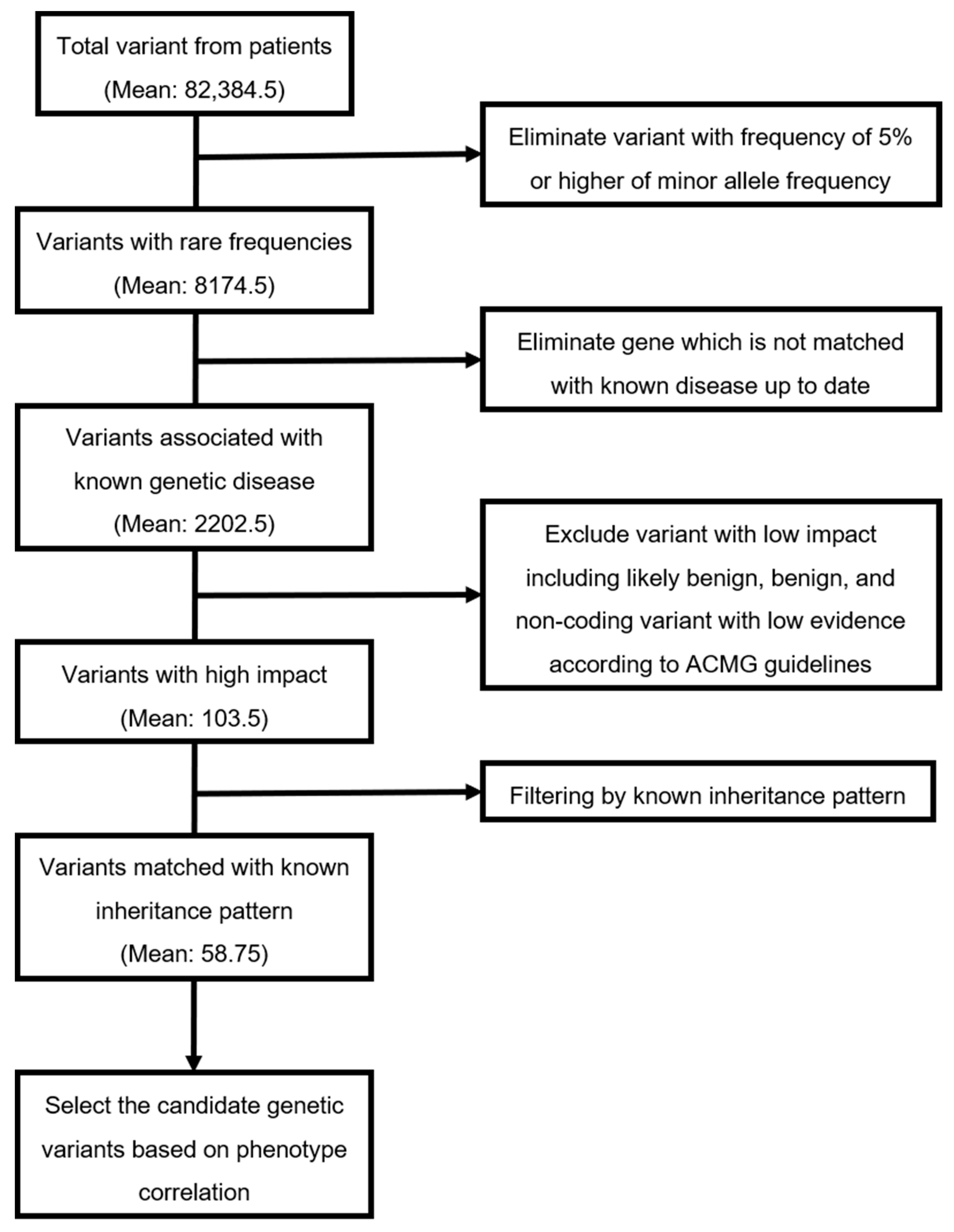
| Average number of variants from patients | 106,311 | 107,509 | 107,600 | 8118 | 82,384.5 |
| Average number of variants with rare frequencies (MAF < 5%) | 10,424 | 10,894 | 10,463 | 917 | 8174.5 |
| Average number of variants associated with known genetic disease | 2737 | 2764 | 2712 | 597 | 2202.5 |
| Average number of variants with high impact | 130 | 95 | 94 | 95 | 103.5 |
| Average number of variants matched with known inheritance pattern | 88 | 46 | 34 | 67 | 58.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-J.; Yum, M.-S.; Seo, G.H.; Lee, Y.; Jang, H.N.; Ko, T.-S.; Lee, B.H. Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders. J. Clin. Med. 2020, 9, 3724. https://doi.org/10.3390/jcm9113724
Kim M-J, Yum M-S, Seo GH, Lee Y, Jang HN, Ko T-S, Lee BH. Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders. Journal of Clinical Medicine. 2020; 9(11):3724. https://doi.org/10.3390/jcm9113724
Chicago/Turabian StyleKim, Min-Jee, Mi-Sun Yum, Go Hun Seo, Yena Lee, Han Na Jang, Tae-Sung Ko, and Beom Hee Lee. 2020. "Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders" Journal of Clinical Medicine 9, no. 11: 3724. https://doi.org/10.3390/jcm9113724
APA StyleKim, M.-J., Yum, M.-S., Seo, G. H., Lee, Y., Jang, H. N., Ko, T.-S., & Lee, B. H. (2020). Clinical Application of Whole Exome Sequencing to Identify Rare but Remediable Neurologic Disorders. Journal of Clinical Medicine, 9(11), 3724. https://doi.org/10.3390/jcm9113724