Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies

 ,
,
Abstract
:1. Introduction
1.1. Dilated Cardiomyopathy: Prevalence, Causes, and Clinical Manifestations
1.2. Dilated Cardiomyopathy in Muscular Dystrophy: Prevalence, Clinical Manifestations
1.3. Objective of This Review
2. Molecular Mechanisms of Dilated Cardiomyopathy
2.1. Genetic and Acquired Causes of DCM
2.1.1. Sarcomere
2.1.2. Cytoskeleton
2.1.3. Acquired Causes of DCM
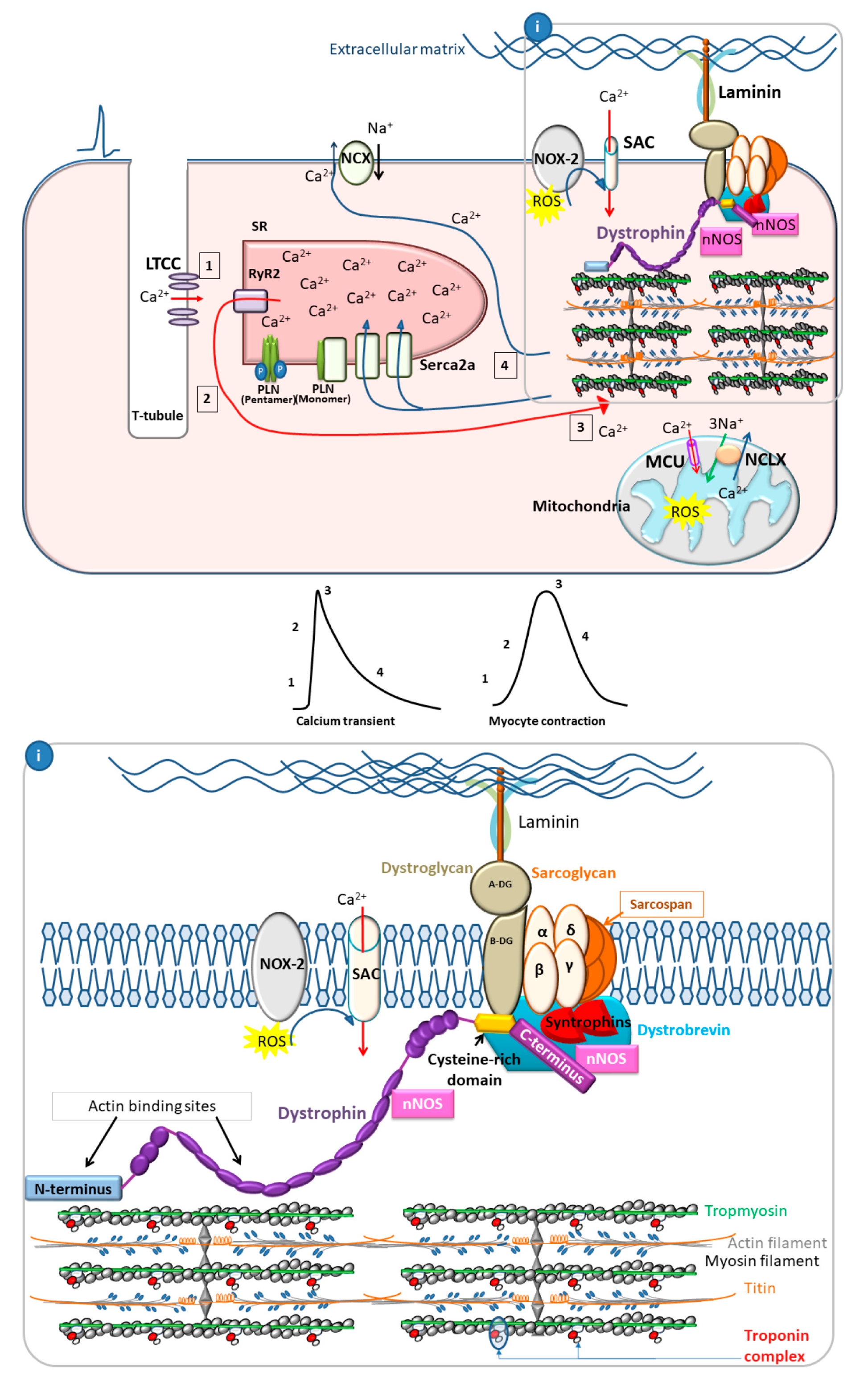
2.2. The Role of Calcium Cycling in DCM Pathogenesis
2.2.1. Calcium Cycling in Healthy Cardiac Myocytes
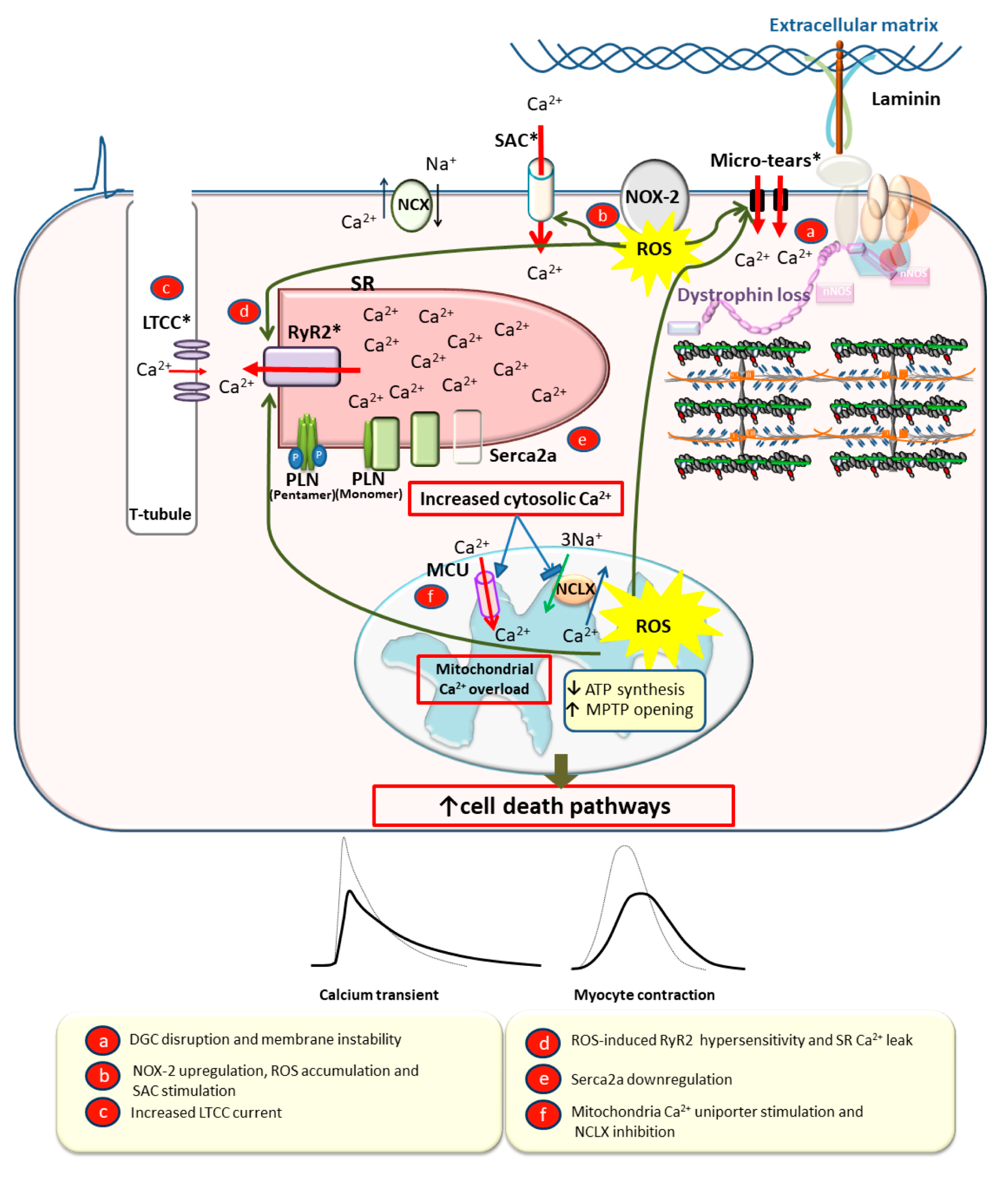
2.2.2. Calcium Cycling Defects in DCM
3. Molecular Mechanisms of DMD Cardiomyopathy
3.1. Dystrophin as a Membrane Stabilizer
3.2. Dystrophin as a Scaffold Protein
3.3. Calcium Overload Leading to Myocyte Death, Fibrosis, and Dilation
4. Model Systems to Study DCM and DMD Cardiomyopathy
4.1. Rodents
4.2. Large Mammals
4.3. Human iPSCs
5. Currently Utilized Therapies for DMD Cardiomyopathy
5.1. Gene Therapy
5.2. Drugs and Small Molecules
6. Experimental Therapeutic Strategies to Improve Calcium Handling and Decrease Calcium Overload in DCM and DMD Cardiomyopathy
6.1. Membrane Stabilization
6.2. Stretch-Activated Channel Inhibition
6.3. RyR2 Stabilization
6.4. Modulation of Serca2a/PLN
6.4.1. Serca2a as a Target for DCM Therapy
6.4.2. PLN as a Target for DCM Therapy
6.4.3. Serca2a and PLN in Muscular Dystrophy-Associated Cardiomyopathy
6.4.4. Caution with Serca2a/PLN Therapy for Cardiomyopathy
6.5. Calcium Buffering
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. World Heath Statistics 2017: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Towbin, J.A. Dilated cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romitti, P.A.; Zhu, Y.; Puzhankara, S.; James, K.A.; Nabukera, S.K.; Zamba, G.K.D.; Ciafaloni, E.; Cunniff, C.; Druschel, C.M.; Mathews, K.D.; et al. Prevalence of duchenne and becker muscular dystrophies in the United States. Pediatrics 2015, 135, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Shen, X.; Dong, X.; Ran, N.; Han, G.; Cao, L.; Gu, B.; Yin, H.F. Peptide nucleic acid promotes systemic dystrophin expression and functional rescue in dystrophin-deficient mdx mice. Mol. Ther. Nucleic Acids 2015, 4, e255. [Google Scholar] [CrossRef]
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Simonds, A.K.; Muntoni, F.; Heather, S.; Fielding, S. Impact of nasal ventilation on survival in hypercapnic Duchenne muscular dystrophy. Thorax 1998, 53, 949–952. [Google Scholar] [CrossRef] [Green Version]
- Porte, P. Clinical indications for noninvasive positive pressure ventilation in chronic respiratory failure due to restrictive lung disease, COPD, and nocturnal hypoventilation—A consensus conference report. Chest 1999, 116, 521–534. [Google Scholar]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J.I. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Podhorska-Okolow, M.; Geromel, V.; Rizzi, C.; Arslan, P.; Franceschi, C.; Carraro, U. Exercise Induces Myonuclear Ubiquitination and Apoptosis in Dystrophin-deficient Muscles of Mice. J. Neuropathol. Exp. Neurol. 1997, 56, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.M.; Wang, D.J.; Shettigar, V.; Roof, S.R.; Canan, B.D.; Bakkar, N.; Shintaku, J.; Gu, J.M.; Little, S.C.; Ratnam, N.M.; et al. NF-κB inhibition rescues cardiac function by remodeling calcium genes in a Duchenne muscular dystrophy model. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Williams, I.A.; Allen, D.G. Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H846–H855. [Google Scholar] [CrossRef] [Green Version]
- Law, M.L.; Prins, K.W.; Olander, M.E.; Metzger, J.M. Exacerbation of dystrophic cardiomyopathy by phospholamban deficiency mediated chronically increased cardiac Ca2+ cycling in vivo. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1544–H1552. [Google Scholar] [CrossRef]
- Fanchaouy, M.; Polakova, E.; Jung, C.; Ogrodnik, J.; Shirokova, N.; Niggli, E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium 2009, 46, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Y.; Zhang, L.; Zhang, X.; Sullivan, R.; Ai, X.; Szeto, C.; Cai, A.; Liu, L.; Xiao, W.; et al. Reduced Myocardial Reserve in Young X-Linked Muscular Dystrophy Mice Diagnosed by Two-Dimensional Strain Analysis Combined with Stress Echocardiography. J. Am. Soc. Echocardiogr. 2017, 30, 815–827. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, S.; Zhang, X.; Li, J.; Ai, X.; Zhang, L.; Yu, D.; Ge, S.; Peng, Y.; Chen, X. Blunted cardiac beta-adrenergic response as an early indication of cardiac dysfunction in Duchenne muscular dystrophy. Cardiovasc. Res. 2014, 103, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcnally, E.M.; Mestroni, L. Dilated cardiomyopathy: Genetic determinants and mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.W.; Yasuda, S.; Chamberlain, J.; Metzger, J.M. Cardiac Consequences to Skeletal Muscle-Centric Therapeutics for Duchenne Muscular Dystrophy. Trends Cardiovasc. Med. 2009, 19, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Berko, B.A.; Swift, M. X-Linked Dialted Cardiomyopathy. N. Engl. J. Med. 1987, 316, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.B.; Swift, M. X-linked dilated cardiomyopathy: Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, M.; Bourke, J.; Bullock, R.; Gibson, M.; Mehta, J.; Giddings, D.; Straub, V.; Bushby, K. Managing Duchenne muscular dystrophy The additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul. Disord. 2007, 17, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of Contraction in Striated Muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Kamisago, M.; Sapna, S.; DePalma, S.; Solomon, S.; Sharma, P.; McDonough, B.; Smoot, L.; Mullen, M.P.; Woolf, P.K.; Wigle, D.; et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 2000, 343, 1688–1696. [Google Scholar] [CrossRef]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.V.; Potter, J.D. Molecular and Celluar Aspects of Troponin Cardiomyopathies. Ann. N. Y. Acad. Sci. 2004, 1015, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.R.; Buck, S.H.; Stoker, S.W.; Greaser, M.L.; Mentzer, R.M. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: Role of altered β-adrenergically mediated protein phosphorylation. J. Clin. Investig. 1996, 98, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, S.J.; Walker, J.S.; Walker, L.A.; Li, X.; Du, Y.; Miyamoto, S.D.; Sucharov, C.C.; Garcia, A.M.; Mitchell, M.B.; Ambardekar, A.V.; et al. Increased myocyte calcium sensitivity in end-stage pediatric dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1221–H1230. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, J.; De Jong, J.W.; Owen, V.J.; Burton, P.B.J.; Stienen, G.J.M. Effect of protein kinase A on calcium sensitivity of force and its sarcomere length dependence in human cardiomyocytes. Cardiovasc. Res. 2000, 46, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Memo, M.; Leung, M.C.; Ward, D.G.; Dos Remedios, C.; Morimoto, S.; Zhang, L.; Ravenscroft, G.; McNamara, E.; Nowak, K.J.; Marston, S.B.; et al. Familial dilated cardiomyopathy mutations uncouple troponin i phosphorylation from changes in myofibrillar Ca2+ sensitivity. Cardiovasc. Res. 2013, 99, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messer, A.E.; Marston, S.B. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 2014, 5, 315. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.N.; Potter, J.D. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail. Rev. 2005, 10, 225–235. [Google Scholar] [CrossRef]
- Mestroni, L.; Brun, F.; Spezzacatene, A.; Sinagra, G.; Taylor, M.R. Genetic Causes of Dilated Cardiomyopathy. Prog. Pediatr Cardiol 2014, 37, 13–18. [Google Scholar] [CrossRef] [Green Version]
- McNally, E.M. Broken giant linked to heart failure. Nature 2012, 483, 281–282. [Google Scholar] [CrossRef]
- Yasuda, S.; Townsend, D.W.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef]
- Barresi, R.; Di Blasi, C.; Tiziana, N.; Brugnoni, R.; Vitali, A.; Felisari, G.; Salandi, A.; Daniel, S.; Cornelio, F.; Morandi, L.; et al. Disruption of heart sarcoglycan complex and severe cardiomyopathy caused by β sarcoglycan mutations. J. Med. Genet. 2000, 37, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Investig. 2000, 106, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.; Elliott, P.; McKenna, W.J. Dilated cardiomyopathy: A genetically heterogeneous disease. Lancet 2002, 360, 654–655. [Google Scholar] [CrossRef]
- Sequeira, V.; Nijenkamp, L.L.A.M.; Regan, J.A.; Van Der Velden, J. The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochim. Biophys. Acta Biomembr. 2014, 1838, 700–722. [Google Scholar] [CrossRef] [Green Version]
- Heydemann, A.; McNally, E.M. Consequences of Disrupting the Dystrophin-Sarcoglycan Complex in Cardiac and Skeletal Myopathy. Trends Cardiovasc. Med. 2007, 17, 55–59. [Google Scholar] [CrossRef]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Kearney, M.T.; Cotton, J.M.; Richardson, P.J.; Shah, A.M. Viral myocarditis and dilated cardiomyopathy: Mechanisms, manifestations, and management. Postgrad. Med. J. 2001, 77, 4–10. [Google Scholar] [CrossRef]
- Barnabei, M.S.; Sjaastad, F.V.; Townsend, D.W.; Bedada, F.B.; Metzger, J.M. Severe dystrophic cardiomyopathy caused by the enteroviral protease 2A-mediated C-terminal dystrophin cleavage fragment. Sci. Transl. Med. 2015, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Parodi, O.; De Maria, R.; Oltrona, L.; Testa, R.; Sambuceti, G.; Roghi, A.; Merli, M.; Belingheri, L.; Accinni, R.; Spinelli, F.; et al. Myocardial blood flow distribution in patients with ischemic heart disease or dilated cardiomyopathy undergoing heart transplantation. Circulation 1993, 88, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Van Den Heuvel, A.F.M.; Van Veldhuisen, D.J.; Van Der Wall, E.E.; Blanksma, P.K.; Siebelink, H.M.J.; Vaalburg, W.M.; Van Gilst, W.H.; Crijns, H.J.G.M. Regional myocardial blood flow reserve impairment and metabolic changes suggesting myocardial ischemia in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2000, 35, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Brittsan, A.G.; Kranias, E.G. Phospholamban and cardiac contractile function. J. Mol. Cell. Cardiol. 2000, 32, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLennan, D.H.; Abu-Abed, M.; Kang, C.H. Structure-function relationships in Ca2+ cycling proteins. J. Mol. Cell. Cardiol. 2002, 34, 897–918. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Limas, C.J.; Olivari, M.T.; Goldenberg, I.F.; Levine, T.B.; Benditt, D.G.; Simon, A. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc. Res. 1987, 21, 601–605. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Dash, R.; Frank, K.F.; Carr, A.N.; Moravec, C.S.; Kranias, E.G. Gender influences on sarcoplasmic reticulum Ca2+-handling in failing human myocardium. J. Mol. Cell. Cardiol. 2001, 33, 1345–1353. [Google Scholar] [CrossRef]
- Meyer, M.; Schillinger, W.; Pieske, B.; Holubarsch, C.; Heilmann, C.; Posival, H.; Kuwajima, G.; Mikoshiba, K.; Just, H.; Hasenfuss, G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 1995, 92, 778–784. [Google Scholar] [CrossRef]
- Townsend, D.W.; Yasuda, S.; Metzger, J. Cardiomyopathy of Duchenne muscular dystrophy: Pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev. Cardiovasc. Ther. 2007, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claflin, D.R.; Brooks, S.V. Direct observation of failing fibers in muscles of dystrophic mice provides mechanistic insight into muscular dystrophy. Am. J. Physiol. Cell Physiol. 2008, 294, C651–C658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.G.; Gervasio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010, 88, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Des Rosiers, C.; Petrof, B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef]
- Townsend, D.; Turner, I.; Yasuda, S.; Martindale, J.; Davis, J.; Shillingford, M.; Kornegay, J.N.; Metzger, J.M. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Investig. 2010, 120, 1140–1150. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Jonathan Lederer, W. X-ROS signalling is enhanced and graded by cyclic cardiomyocyte stretch. Cardiovasc. Res. 2013, 98, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.A.; Allen, D.G. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1969–H1977. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, N.D.; Fanchaouy, M.; Gusev, K.; Shirokova, N.; Niggli, E. Hypersensitivity of excitation-contraction coupling in dystrophic cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1992–H2003. [Google Scholar] [CrossRef]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, K.W.; Asp, M.L.; Zhang, H.; Wang, W.; Metzger, J.M. Microtubule-Mediated Misregulation of Junctophilin-2 Underlies T-Tubule Disruptions and Calcium Mishandling in mdx Mice. JACC Basic Transl. Sci. 2016, 1, 122–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, X.; Rubi, L.; Obermair, G.J.; Cervenka, R.; Dang, X.B.; Lukacs, P.; Kummer, S.; Bittner, R.E.; Kubista, H.; Todt, H.; et al. Enhanced currents through L-type calcium channels in cardiomyocytes disturb the electrophysiology of the dystrophic heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H564–H573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valladares, D.; Almarza, G.; Contreras, A.; Pavez, M.; Buvinic, S.; Jaimovich, E.; Casas, M. Electrical stimuli are anti-apoptotic in skeletal muscle via extracellular ATP. Alteration of this signal in Mdx mice is a likely cause of dystrophy. PLoS ONE 2013, 8, e75340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidlmayer, L.K.; Kuhn, J.; Berbner, A.; Arias-Loza, P.A.; Williams, T.; Kaspar, M.; Czolbe, M.; Kwong, J.Q.; Molkentin, J.D.; Heinze, K.G.; et al. Inositol 1,4,5-Trisphosphate-mediated sarcoplasmic reticulum-mitochondrial crosstalk influences adenosine triphosphate production via mitochondrial Ca2+ uptake through the mitochondrial ryanodine receptor in cardiac myocytes. Cardiovasc. Res. 2016, 112, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, X.; Kubo, H.; Harris, D.M.; Mills, G.D.; Moyer, J.; Berretta, R.; Potts, S.T.; Marsh, J.D.; Houser, S.R. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 2005, 97, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; Van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Viola, H.M.; Adams, A.M.; Davies, S.M.K.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef] [Green Version]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1113–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J.; Stöllberger, C. The heart in human dystrophinopathies. Cardiology 2003, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Connuck, D.M.; Sleeper, L.A.; Colan, S.D.; Cox, G.F.; Towbin, J.A.; Lowe, A.M.; Wilkinson, J.D.; Orav, E.J.; Cuniberti, L.; Salbert, B.A.; et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: A comparative study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 2008, 155, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Cox, G.F.; Kunkel, L.M. Dystrophies and heart disease. Curr. Opin. Cardiol. 1997, 12, 329–343. [Google Scholar] [CrossRef]
- Frankel, K.A.; Rosser, R.J. The pathology of the heart in progressive muscular dystrophy: Epimyocardial fibrosis. Hum. Pathol. 1976, 7, 375–386. [Google Scholar] [CrossRef]
- Giglio, V.; Pasceri, V.; Messano, L.; Mangiola, F.; Pasquini, L.; Dello Russo, A.; Damiani, A.; Mirabella, M.; Galluzzi, G.; Tonali, P.; et al. Ultrasound tissue characterization detects preclinical myocardial structural changes in children affected by Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2003, 42, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Sakata, K.; Kachi, E.; Hirata, S.; Ishihara, T.; Ishikawa, K. Sequential changes in cardiac structure and function in patients with Duchenne type muscular dystrophy: A two-dimensional echocardiographic study. Am. Heart J. 1998, 135, 937–944. [Google Scholar] [CrossRef]
- Takenaka, A.; Yokota, M.; Iwase, M.; Miyaguchi, K.; Hayashi, H.; Saito, H. Discrepancy between systolic and diastolic dysfunction of the left ventricle in patients with Duchenne muscular dystrophy. Eur. Heart J. 1993, 14, 669–676. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ross, J. Models of dilated cardiomyopathy in the mouse and the hamster. Curr. Opin. Cardiol. 2000, 15, 197–201. [Google Scholar] [CrossRef]
- Arber, S.; Hunter, J.J.; Ross, J.; Hongo, M.; Sansig, G.; Borg, J.; Perriard, J.C.; Chien, K.R.; Caroni, P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 1997, 88, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, M.B.; Bauer, R.; Jungmann, A.; Winter, L.; Rapti, K.; Strucksberg, K.H.; Clemen, C.S.; Li, Z.; Schröder, R.; Katus, H.A.; et al. AAV9-mediated gene transfer of desmin ameliorates cardiomyopathy in desmin-deficient mice. Gene Ther. 2016, 23, 673–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Yuan, F.; Mu, J.; Li, C.; Chen, N.; Guo, S.; Kingery, J.; Prabhu, S.D.; Bolli, R.; Rokosh, G. Chronic AMD3100 antagonism of SDF-1α-CXCR4 exacerbates cardiac dysfunction and remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 2010, 49, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, L.H.; Entman, M.L.; Hartley, C.J.; Youker, K.A.; Zhu, J.; Hall, S.R.; Hawkins, H.K.; Berens, K.; Ballantyne, C.M. Myocardial ischemia and reperfusion: A murine model. Am. J. Physiol. Heart Circ. Physiol. 1995, 269, H2147–H2154. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.M.; Mihm, M.J.; Bauer, J.A. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J. Pharmacol. Exp. Ther. 2000, 294, 396–401. [Google Scholar]
- Robert, J. Long-term and short-term models for studying anthracycline cardiotoxicity and protectors. Cardiovasc. Toxicol. 2007, 7, 135–139. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Pfeffer, J.M.; Pfeffer, M.A. Progressive ventricular remodeling in response to diffuse isoproterenol-induced myocardial necrosis in rats. Circ. Res. 1994, 75, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Crackower, M.A.; Eriksson, U.; Sarao, R.; Kozieradzki, I.; Sasaki, T.; Irie-Sasaki, J.; Gidrewicz, D.; Rybin, V.O.; Wada, T.; et al. Phosphoinositide 3-Kinase γ-Deficient Mice Are Protected From Isoproterenol-Induced Heart Failure. Circulation 2003, 108, 2147–2152. [Google Scholar] [CrossRef] [Green Version]
- Hainsey, T.A.; Senapati, S.; Kuhn, D.E.; Rafael, J.A. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul. Disord. 2003, 13, 294–302. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Hiranandani, N.; Mays, T.A.; Rafael-Fortney, J.A. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 2373–2378. [Google Scholar] [CrossRef] [Green Version]
- Grady, R.M.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanest, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Hearse, D.J.; Sutherland, F.J. Experimental Models for the Study of Cardiovascular Function and Disease Defining the Question and Identifying the Model. Pharmacol. Res. 2000, 41, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, J.; Mathison, M.; Tondato, F.; Mulkey, S.P.; Micko, C.; Chronos, N.A.F.; Robinson, K.A. A clinically relevant large-animal model for evaluation of tissue-engineered cardiac surgical patch materials. Cardiovasc. Revasc. Med. 2005, 6, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Jugdutt, B.I.; Jelani, A.; Palaniyappan, A.; Idikio, H.; Uweira, R.E.; Menon, V.; Jugdutt, C.E. Aging-related early changes in markers of ventricular and matrix remodeling after reperfused st-segment elevation myocardial infarction in the canine model: Effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation 2010, 122, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; White, A.J.; Matsushita, S.; Malliaras, K.; Steenbergen, C.; Zhang, Y.; Li, T.S.; Terrovitis, J.; Yee, K.; Simsir, S.; et al. Intramyocardial injection of autologous cardiospheres or cardiosphere-derived cells preserves function and minimizes adverse ventricular remodeling in pigs with heart failure post-myocardial infarction. J. Am. Coll. Cardiol. 2011, 57, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Gorman, J.H.; Gorman, R.C.; Plappert, T.; Jackson, B.M.; Hiramatsu, Y.; St. John-Sutton, M.G.; Edmunds, J. Infarct size and location determine development of mitral regurgitation in the sheep model. J. Thorac. Cardiovasc. Surg. 1998, 115, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthélémy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kuhr, C.S.; Allen, J.M.; Blankinship, M.; Gregorevic, P.; Chamberlain, J.S.; Tapscott, S.J.; Storb, R. Sustained AAV-mediated dystrophin expression in a canine model of duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 2007, 15, 1160–1166. [Google Scholar] [CrossRef]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef] [Green Version]
- Siu, C.W.; Lee, Y.K.; Ho, J.C.Y.; Lai, W.H.; Chan, Y.C.; Ng, K.M.; Wong, L.Y.; Au, K.W.; Lau, Y.M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging Albany NY 2012, 4, 803–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, H.F.; Ho, J.C.Y.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Li, Y.; Han, L.; Kaplan, A.D.; Ao, Y.; Kalra, S.; Bett, G.C.L.; Rasmusson, R.L.; Denning, C.; Yang, L. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with duchenne muscular dystrophy. DMM Dis. Model. Mech. 2015, 8, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recchia, F.A.; Lionetti, V. Animal models of dilated cardiomyopathy for translational research. Vet. Res. Commun. 2007, 31, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.; Haghighi, K.; Kranias, E.G. From mouse to man: Understanding heart failure through genetically altered mouse models. J. Cardiac Fail. 2002, 8, S432–S449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.D.; Bohlooly, M.; Sjöquist, P.O. Murine models for the study of congestive heart failure: Implications for understanding molecular mechanisms and for drug discovery. J. Pharmacol. Toxicol. Methods 2004, 50, 163–174. [Google Scholar] [CrossRef]
- Grady, R.M.; Grange, R.W.; Lau, K.S.; Maimone, M.M.; Nichol, M.C.; Stull, J.T.; Sanes, J.R. Role for α-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell Biol. 1999, 1, 215–220. [Google Scholar] [CrossRef]
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Rader, E.P.; Levy, J.R.; Bansal, D.; Campbell, K.P. Dystrophin deficiency exacerbates skeletal muscle pathology in dysferlin-null mice. Skelet. Muscle 2011, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Hosur, V.; Kavirayani, A.; Riefler, J.; Carney, L.M.B.; Lyons, B.; Gott, B.; Cox, G.A.; Shultz, L.D. Dystrophin and dysferlin double mutant mice: A novel model for rhabdomyosarcoma. Cancer Genet. 2012, 205, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Banks, G.B.; Combs, A.C.; Odom, G.L.; Bloch, R.J.; Chamberlain, J.S. Muscle Structure Influences Utrophin Expression in mdx Mice. PLoS Genet. 2014, 10, e1004431. [Google Scholar] [CrossRef] [Green Version]
- Gawlik, K.I.; Holmberg, J.; Durbeej, M. Loss of dystrophin and β-sarcoglycan significantly exacerbates the phenotype of laminin α2 chain-deficient animals. Am. J. Pathol. 2014, 184, 740–752. [Google Scholar] [CrossRef]
- Monnet, E.; Chachques, J.C. Animal models of heart failure: What is new? Ann. Thorac. Surg. 2005, 79, 1445–1453. [Google Scholar] [CrossRef]
- Dixon, J.A.; Spinale, F.G. Large animal models of heart failure; A critical link in the translation of basic science to clinical practice. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [CrossRef] [Green Version]
- Camacho, P.; Fan, H.; Liu, Z.; He, J.Q. Small mammalian animal models of heart disease. Am. J. Cardiovasc. Dis. 2016, 6, 70–80. [Google Scholar]
- Valentine, B.A.; Cummings, J.F.; Cooper, B.J. Development of Duchenne-type cardiomyopathy. Morphologic studies in a canine model. Am. J. Pathol. 1989, 135, 671–678. [Google Scholar]
- Moise, N.S.; Valentine, B.A.; Brown, C.A.; Erb, H.N.; Beck, K.A.; Cooper, B.J.; Gilmour, R.F. Duchenne’s cardiomyopathy in a canine model: Electrocardiographic and echocardiographic studies. J. Am. Coll. Cardiol. 1991, 17, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Devaux, J.Y.; Cabane, L.; Esler, M.; Flaouters, H.; Duboc, D. Non-invasive evaluation of the cardiac function in Golden Retriever dogs by radionuclide angiography. Neuromuscul. Disord. 1993, 3, 429–432. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Mack, D.L.; Moreno, C.M.; Strande, J.L.; Mathieu, J.; Shi, Y.; Markert, C.D.; Wang, Z.; Liu, G.; Lawlor, M.W.; et al. Dystrophin-deficient cardiomyocytes derived from human urine: New biologic reagents for drug discovery. Stem Cell Res. 2014, 12, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Tsurumi, F.; Baba, S.; Yoshinaga, D.; Umeda, K.; Hirata, T.; Takita, J.; Heike, T. The intracellular Ca2+ concentration is elevated in cardiomyocytes differentiated from hiPSCs derived from a Duchenne muscular dystrophy patient. PLoS ONE 2019, 14, e0213768. [Google Scholar] [CrossRef] [Green Version]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Griggs, R.C.; Moxley, R.T.; Mendell, J.R.; Fenichel, G.M.; Brooke, M.H.; Pestronk, A.; Miller, J.P. Prednisone in Duchenne Dystrophy: A Randomized, Controlled Trial Defining the Time Course and Dose Response. Arch. Neurol. 1991, 48, 383–388. [Google Scholar] [CrossRef]
- Biggar, W.D.; Harris, V.A.; Eliasoph, L.; Alman, B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord. 2006, 16, 249–255. [Google Scholar] [CrossRef]
- Spurney, C.F. Cardiomyopathy of duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve 2011, 44, 8–19. [Google Scholar] [CrossRef]
- Schram, G.; Fournier, A.; Leduc, H.; Dahdah, N.; Therien, J.; Vanasse, M.; Khairy, P. All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2013, 61, 948–954. [Google Scholar] [CrossRef]
- Raman, S.V.; Cripe, L.H. Glucocorticoid therapy for Duchenne cardiomyopathy: A Hobson’s choice? J. Am. Heart Assoc. 2015, 4, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Barber, B.J.; Andrews, J.G.; Lu, Z.; West, N.A.; Meaney, F.J.; Price, E.T.; Gray, A.; Sheehan, D.W.; Pandya, S.; Yang, M.; et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J. Pediatr. 2013, 163, 1080–1084. [Google Scholar] [CrossRef]
- Griggs, R.C.; Herr, B.E.; Reha, A.; Elfring, G.; Atkinson, L.; Cwik, V.; Mccoll, E.; Tawil, R.; Pandya, S.; Mcdermott, M.P.; et al. Corticosteroids in Duchenne muscular dystrophy: Major variations in practice. Muscle Nerve 2013, 48, 27–31. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Reeves, E.; Damsker, J.; Nagaraju, K.; McCall, J.M.; Connor, E.M.; Bushby, K. Novel Approaches to Corticosteroid Treatment in Duchenne Muscular Dystrophy. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.; Straub, V.; Blain, A.; Bushby, K.; MacGowan, G.A. Contrasting effects of steroids and angiotensin-converting-enzyme inhibitors in a mouse model of dystrophin-deficient cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 463–471. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Murray, J.D.; Schill, K.E.; Rastogi, N.; Schultz, E.J.; Tran, T.; Raman, S.V.; Rafael-Fortney, J.A. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of duchenne muscular dystrophy. PLoS ONE 2014, 9, e88360. [Google Scholar] [CrossRef]
- Ogata, H.; Ishikawa, Y.; Ishikawa, Y.; Minami, R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J. Cardiol. 2009, 53, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Dasgupta, C.; Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov. Today 2011, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Forrester, S.J.; O’Brian, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125, 4–13. [Google Scholar] [CrossRef]
- Bernal, J.; Pitta, S.R.; Thatai, D. Role of the renin-angiotensin-aldosterone system in diastolic heart failure: Potential for pharmacologic intervention. Am. J. Cardiovasc. Drugs 2006, 6, 373–381. [Google Scholar] [CrossRef]
- Duboc, D.; Meune, C.; Pierre, B.; Wahbi, K.; Eymard, B.; Toutain, A.; Berard, C.; Vaksmann, G.; Weber, S.; Bécane, H.M. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years’ follow-up. Am. Heart J. 2007, 154, 596–602. [Google Scholar] [CrossRef]
- Bangalore, S.; Fakheri, R.; Toklu, B.; Ogedegbe, G.; Weintraub, H.; Messerli, F.H. Angiotensin-Converting Enzyme Inhibitors or Angiotensin Receptor Blockers in Patients Without Heart Failure? Insights from 254,301 Patients from Randomized Trials. Mayo Clin. Proc. 2016, 91, 51–60. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The Sympathetic Nervous System in Heart Failure. Physiology, Pathophysiology, and Clinical Implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [Green Version]
- Klapholz, M. Β-Blocker Use for the Stages of Heart Failure. Mayo Clin. Proc. 2009, 84, 718–729. [Google Scholar] [CrossRef] [Green Version]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.B.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [Green Version]
- Kajimoto, H.; Ishigaki, K.; Okumura, K.; Tomimatsu, H.; Nakazawa, M.; Saito, K.; Osawa, M.; Nakanishi, T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ. J. 2006, 70, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, T.; Tamura, T.; Kuru, S.; Kikuchi, Y.; Kawai, M. Carvedilol can prevent cardiac events in duchenne muscular dystrophy. Intern. Med. 2010, 49, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Halnon, N.J.; Kissel, J.T.; He, X.; Tran, T.; Smart, S.; McCarthy, B.; Taylor, M.D.; et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Spurney, C.F.; Guerron, A.D.; Yu, Q.; Sali, A.; van der Meulen, J.H.; Hoffman, E.P.; Nagaraju, K. Membrane Sealant Poloxamer P188 Protects Against Isoproterenol Induced Cardiomyopathy in Dystrophin Deficient Mice. BMC Cardiovasc. Disord. 2011, 11, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.J.; Metzger, J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J. Mol. Cell. Cardiol. 2014, 67, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.L.; Williams, I.A.; Chu, Y.; Cooper, P.J.; Ju, Y.K.; Allen, D.G. Stretch-activated channels in the heart: Contributions to length-dependence and to cardiomyopathy. Prog. Biophys. Mol. Biol. 2008, 97, 232–249. [Google Scholar] [CrossRef]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Bostick, B.; Yue, Y.; Hajjar, R.; Duan, D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J. Transl. Med. 2011, 9, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Hoshijima, M.; Ikeda, Y.; Iwanaga, Y.; Minamisawa, S.; Date, M.O.; Gu, Y.; Iwatate, M.; Li, M.; Wang, L.; Wilson, J.M.; et al. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat. Med. 2002, 8, 864–871. [Google Scholar] [CrossRef]
- Dieterle, T.; Meyer, M.; Gu, Y.; Belke, D.D.; Swanson, E.; Iwatate, M.; Hollander, J.; Peterson, K.L.; Ross, J.; Dillmann, W.H. Gene transfer of a phospholamban-targeted antibody improves calcium handling and cardiac function in heart failure. Cardiovasc. Res. 2005, 67, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Ikeda, Y.; Yano, M.; Yoshimura, K.; Nishino, S.; Aoyama, H.; Wang, L.; Aoki, H.; Matsuzaki, M. Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J. 2006, 20, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Houang, E.M.; Bartos, J.; Hackel, B.J.; Lodge, T.P.; Yannopoulos, D.; Bates, F.S.; Metzger, J.M. Cardiac Muscle Membrane Stabilization in Myocardial Reperfusion Injury. JACC Basic Transl. Sci. 2019, 4, 275–287. [Google Scholar] [CrossRef]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef] [Green Version]
- Vandebrouck, C.; Martin, D.; Van Schoor, M.C.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- Huang, F.; Shan, J.; Reiken, S.; Wehrens, X.H.T.; Marks, A.R. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: Rescue of heart failure by calstabin2 in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3456–3461. [Google Scholar] [CrossRef] [Green Version]
- Capogrosso, R.F.; Mantuano, P.; Uaesoontrachoon, K.; Cozzoli, A.; Giustino, A.; Dow, T.; Srinivassane, S.; Filipovic, M.; Bell, C.; Vandermeulen, J.; et al. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: Proof-of-concept study and independent validation of efficacy. FASEB J. 2018, 32, 1025–1043. [Google Scholar] [CrossRef] [Green Version]
- Del Monte, F.; Harding, S.E.; Schmidt, U.; Matsui, T.; Kang, Z.B.; Dec, G.W.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999, 100, 2308–2311. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, M.I.; Del Monte, F.; Schmidt, U.; DiSalvo, T.S.; Kang, Z.B.; Matsui, T.; Guerrero, J.L.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Adenoviral gene transfer of SERCA2A improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc. Natl. Acad. Sci. USA 2000, 97, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Del Monte, F.; Williams, E.; Lebeche, D.; Schmidt, U.; Rosenzweig, A.; Gwathmey, J.K.; Lewandowski, E.D.; Hajjar, R.J. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca2+-ATPase in a rat model of heart failure. Circulation 2001, 104, 1424–1429. [Google Scholar] [CrossRef] [Green Version]
- Sakata, S.; Lebeche, D.; Sakata, N.; Sakata, Y.; Chemaly, E.R.; Liang, L.F.; Tsuji, T.; Takewa, Y.; del Monte, F.; Peluso, R.; et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J. Mol. Cell. Cardiol. 2007, 42, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Niwano, K.; Arai, M.; Koitabashi, N.; Watanabe, A.; Ikeda, Y.; Miyoshi, H.; Kurabayashi, M. Lentiviral vector-mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial Infarction in rats. Mol. Ther. 2008, 16, 1026–1032. [Google Scholar] [CrossRef]
- Byrne, M.J.; Power, J.M.; Preovolos, A.; Mariani, J.A.; Hajjar, R.J.; Kaye, D.M. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008, 15, 1550–1557. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.A.; Smolic, A.; Preovolos, A.; Byrne, M.J.; Power, J.M.; Kaye, D.M. Augmentation of left ventricular mechanics by recirculation-mediated AAV2/1SERCA2a gene delivery in experimental heart failure. Eur. J. Heart Fail. 2011, 13, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Jessup, M.; Greenberg, B.; Mancini, D.; Cappola, T.; Pauly, D.F.; Jaski, B.; Yaroshinsky, A.; Zsebo, K.M.; Dittrich, H.; Hajjar, R.J. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): A phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011, 124, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Zsebo, K.; Yaroshinsky, A.; Rudy, J.J.; Wagner, K.; Greenberg, B.; Jessup, M.; Hajjar, R.J. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: Analysis of recurrent cardiovascular events and mortality. Circ. Res. 2014, 114, 101–108. [Google Scholar] [CrossRef]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Kadambi, V.J.; Ponniah, S.; Harrer, J.M.; Hoit, B.D.; Dorn, G.W.; Walsh, R.A.; Kranias, E.G. Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J. Clin. Investig. 1996, 97, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Grupp, I.L.; Harrer, J.; Ponniah, S.; Grupp, G.; Duffy, J.J.; Doetschman, T.; Kranias, E.G. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of β-agonist stimulation. Circ. Res. 1994, 75, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Wolska, B.M.; Grupp, I.L.; Harrer, J.M.; Haghighi, K.; Ferguson, D.G.; Slack, J.P.; Grupp, G.; Doetschman, T.; Solaro, R.J.; et al. Phospholamban gene dosage effects in the mammalian heart. Circ. Res. 1996, 78, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Minamisawa, S.; Hoshijima, M.; Chu, G.; Ward, C.A.; Frank, K.; Gu, Y.; Martone, M.E.; Wang, Y.; Ross, J.; Kranias, E.G.; et al. Chronic phospholamban-sarcoplasmic reticulum calcium atpase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell 1999, 99, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Iwanaga, Y.; Hoshijima, M.; Gu, Y.; Iwatate, M.; Dieterle, T.; Ikeda, Y.; Date, M.O.; Chrast, J.; Matsuzaki, M.; Peterson, K.L.; et al. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J. Clin. Investig. 2004, 113, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Kaye, D.M.; Preovolos, A.; Marshall, T.; Byrne, M.; Hoshijima, M.; Hajjar, R.; Mariani, J.A.; Pepe, S.; Chien, K.R.; Power, J.M. Percutaneous Cardiac Recirculation-Mediated Gene Transfer of an Inhibitory Phospholamban Peptide Reverses Advanced Heart Failure in Large Animals. J. Am. Coll. Cardiol. 2007, 50, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, Y.; Ikeda, Y.; Shiraishi, K.; Fujimoto, S.N.; Aoyama, H.; Yoshimura, K.; Inui, M.; Hoshijima, M.; Kasahara, H.; Aoki, H.; et al. Heart failure-inducible gene therapy targeting protein phosphatase 1 prevents progressive left ventricular remodeling. PLoS ONE 2012, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rohman, M.S.; Emoto, N.; Takeshima, Y.; Yokoyama, M.; Matsuo, M. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem. Biophys. Res. Commun. 2003, 310, 228–235. [Google Scholar] [CrossRef]
- Goonasekera, S.A.; Lam, C.K.; Millay, D.P.; Sargent, M.A.; Hajjar, R.J.; Kranias, E.G.; Molkentin, J.D. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Investig. 2011, 121, 1044–1052. [Google Scholar] [CrossRef] [Green Version]
- Mázala, D.A.G.; Pratt, S.J.P.; Chen, D.; Molkentin, J.D.; Lovering, R.M.; Chin, E.R. SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol. 2015, 308, C699–C709. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Martone, M.; Gu, Y.; Hoshijima, M.; Thor, A.; Oh, S.S.; Peterson, K.L.; Ross, J. Altered membrane proteins and permeability correlate with cardiac dysfunction in cardiomyopathic hamsters. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1362–H1370. [Google Scholar] [CrossRef] [Green Version]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, G.; Luo, W.; Slack, J.P.; Tilgmann, C.; Sweet, W.E.; Spindler, M.; Saupe, K.W.; Boivin, G.P.; Moravec, C.S.; Matlib, M.A.; et al. Compensatory mechanisms associated with the hyperdynamic function of phospholamban-deficient mouse hearts. Circ. Res. 1996, 79, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Pater, L.; Lynch, R.A.; Asahi, M.; Gramolini, A.O.; Fan, G.C.; Tsiapras, D.; Hahn, H.S.; Adamopoulos, S.; et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Investig. 2003, 111, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Hua Li, G.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef]
- Ha, K.N.; Mastersona, L.R.; Hou, Z.; Verardi, R.; Walsh, N.; Veglia, G.; Robia, S.L. Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc. Natl. Acad. Sci. USA 2011, 108, 2735–2740. [Google Scholar] [CrossRef] [Green Version]
- Abrol, N.; De Tombe, P.P.; Robia, S.L. Acute inotropic and lusitropic effects of cardiomyopathic R9C mutation of phospholamban. J. Biol. Chem. 2015, 290, 7130–7140. [Google Scholar] [CrossRef] [Green Version]
- Small, K.M.; Wagoner, L.E.; Levin, A.M.; Kardia, S.L.R.; Liggett, S.B. Synergistic polymorphisms of β 1- and α 2C-adrenergic receptors and the risk of congestive heart failure. N. Engl. J. Med. 2002, 347, 1135–1142. [Google Scholar] [CrossRef]
- Haghighi, K.; Gregory, K.N.; Kranias, E.G. Sarcoplasmic reticulum Ca-ATPase-phospholamban interactions and dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2004, 322, 1214–1222. [Google Scholar] [CrossRef]
- Permyakov, E.A. Parvalbumin; Uversky, V.N., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2007. [Google Scholar]
- Hapak, R.C.; Zhao, H.; Boschi, J.M.; Henzl, M.T. Novel avian thymic parvalbumin displays high degree of sequence homology to oncomodulin. J. Biol. Chem. 1994, 269, 5288–5296. [Google Scholar]
- Pauls, T.L.; Cox, J.A.; Berchtold, M.W. The Ca2+-binding proteins parvalbumin and oncomodulin and their genes: New structural and functional findings. Biochim. Biophys. Acta Gene Struct. Expr. 1996, 1306, 39–54. [Google Scholar] [CrossRef]
- Falke, J.J.; Drake, S.K.; Hazard, A.L.; Peersen, O.B. Molecular Tuning of Ion Binding to Calcium Signaling Proteins. Q. Rev. Biophys. 1994, 27, 219–290. [Google Scholar] [CrossRef]
- Wahr, P.A.; Michele, D.E.; Metzger, J.M. Parvalbumin gene transfer corrects diastolic dysfunction in diseased cardiac myocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 11982–11985. [Google Scholar] [CrossRef] [Green Version]
- Huq, F.; Lebeche, D.; Iyer, V.; Liao, R.; Hajjar, R.J. Gene transfer of parvalbumin improves diastolic dysfunction in senescent myocytes. Circulation 2004, 109, 2780–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenbaugh, D.W.; Wang, W.; Davis, J.; Edwards, T.; Potter, J.D.; Metzger, J.M. Parvalbumin isoforms differentially accelerate cardiac myocyte relaxation kinetics in an animal model of diastolic dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1705–H1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatkowski, M.L.; Westfall, M.V.; Gomez, C.A.; Wahr, P.A.; Michele, D.E.; DelloRusso, C.; Turner, I.I.; Hong, K.E.; Albayya, F.P.; Metzger, J.M. In vivo acceleration of heart relaxation performance by parvalbumin gene delivery. J. Clin. Investig. 2001, 107, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michele, D.E.; Szatkowski, M.L.; Albayya, F.P.; Metzger, J.M. Parvalbumin gene delivery improves diastolic function in the aged myocardium in vivo. Mol. Ther. 2004, 10, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Zhu, X.; Lebeche, D.; Huq, F.; Guerrero, J.L.; Hajjar, R.J. In vivo gene transfer of parvalbumin improves diastolic function in aged rat hearts. Cardiovasc. Res. 2005, 66, 318–323. [Google Scholar] [CrossRef]
- Coutu, P.; Metzger, J.M. Genetic manipulation of calcium-handling proteins in cardiac myocytes. II. Mathematical modeling studies. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H613–H631. [Google Scholar] [CrossRef] [Green Version]
- Coutu, P.; Metzger, J.M. Genetic manipulation of calcium-handling proteins in cardiac myocytes. I. Experimental studies. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H601–H612. [Google Scholar] [CrossRef] [Green Version]
- Coutu, P.; Metzger, J.M. Optimal range for parvalbumin as relaxing agent in adult cardiac myocytes: Gene transfer and mathematical modeling. Biophys. J. 2002, 82, 2565–2579. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Barnabei, M.S.; Asp, M.L.; Heinis, F.I.; Arden, E.; Davis, J.; Braunlin, E.; Li, Q.; Davis, J.P.; Potter, J.D.; et al. Noncanonical EF-hand motif strategically delays Ca2+ buffering to enhance cardiac performance. Nat. Med. 2013, 19, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asp, M.L.; Sjaastad, F.V.; Siddiqui, J.K.; Davis, J.P.; Metzger, J.M. Effects of Modified Parvalbumin EF-Hand Motifs on Cardiac Myocyte Contractile Function. Biophys. J. 2016, 110, 2094–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, R.; Nascimben, L.; Friedrich, J.; Gwathmey, J.K.; Ingwall, J.S. Decreased energy reserve in an animal model of dilated cardiomyopathy: Relationship to contractile performance. Circ. Res. 1996, 78, 893–902. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model System | Strategy | Cardiac Phenotype |
|---|---|---|
| Rodent | ||
| Muscle LIM protein (MLP) null mice [92] | Deletion of MLP (actin-associated cytoskeletal protein) | Anatomical and physiological hallmarks of human DCM |
| Desmin-deficient mice [91,93] | Desmin knockout mice | Severe loss of overall myocardial architecture by degeneration and calcification |
| Surgical interruptions of coronary arteries [94,95] | Produce myocardial infarction through permanent coronary ligation or re-perfused infarction | DCM phenotype progressively develops post-infarction |
| Doxorubicin or isoproterenol [96,97,98,99] | Toxic drug-mediated cardiomyopathy | Dose-dependent dilated phenotype and overt heart failure over time owing to severe myocardial injury and cell death |
| mdx mice [100,101] | Nonsense point mutation in exon 23 preventing dystrophin expression | Moderate DCM and functional cardiac impairment, progressive with age |
| Utrophin knockout mdx mice [102,103] | Crossing mdx mice to the utrophin-null background | Severe cardiomyopathy. Displays physiological indicators of end-stage heart failure |
| Large animals | ||
| Dogs, pigs and sheep [104,105,106,107,108] | Myocardial infarction, coronary micro-embolization, pacing-induced tachycardia, and toxic injury | DCM phenotype progressively develops post-infarction |
| Golden retriever muscular dystrophy (GRMD) animal model of DMD [68,109,110] | Spontaneous splice site mutation in the DMD gene. Single nucleotide change that leads to exon skipping and an out-of-frame DMD transcript. | Prominent cardiac lesions present as early as 6 months of age, with ECG abnormalities present at 1 year and profound myocardial contractile abnormalities by 20 months |
| Human iPSCs | ||
| iPSCs-CMs [111] | iPSCs-CMs derived from a member of a family with DCM carrying a heterozygous mutation in cardiac troponin T | iPSC-derived cardiomyocytes from DCM patients recapitulated to some extent the morphological and functional phenotypes of familial DCM with inherited mutation in troponin T |
| iPSCs-CMs [112] | Patient-specific DCM iPSC generated from a single member of a family with an autosomal dominant nonsense mutation (p.R225X) in exon 4 of the lamin A/C (LMNA) gene | iPSC-CMs showed morphologic changes, including a higher prevalence of nuclear bleb formation, micronucleation, as well as nuclear senescence and cellular apoptosis |
| iPSC-CMs [113] | iPSC-CMs derived from a patient with dilated cardiomyopathy with a novel heterozygous mutation of p.A285V codon conversion on exon 4 of the desmin gene | iPSC-CMs provided histologic and functional confirmation that the candidate gene variant detected by whole exome sequencing was responsible for the disease |
| iPSCs-CMs [114] | iPSC-CMs from DMD patients and healthy control | In vitro model that manifests the major phenotypes of DCM in DMD patients |
| Target | Therapy | Model | Major Findings |
|---|---|---|---|
| Sarcolemma [41,68,158,159] | Copolymer –based membrane stabilizers | mdx mice dysferlin KO mice GRMD canine | ↓ Myocyte Ca2+ influx/hypercontracture ↓ Stress-induced functional deficits (acute and chronic) ↓ Fibrosis, serum cTnI, LV remodeling ↓ Ex vivo ischemia/reperfusion injury |
| Stretch-Activated Channels [160] | GsMTX-4 | mdx mice | ↓ Myocyte resting Ca2+ concentration |
| Ryanodine Receptor [161] | N-acetyl cysteine Rycal S107 | mdx mice | ↓ Myocyte resting Ca2+ concentration ↓ Myocyte RyR2 Ca2+ leak ↓ Arrhythmias |
| Serca2a [162] | AAV-9 Serca2a | mdx mice | Normalized ECG measurements |
| Phospholamban [19,163,164,165] | AAV S16E-PLN Adenovirus anti-PLN antibody Adenovirus inhibitor-2 PLN-KO | BIO14.6 hamster mdx mice | PLN inhibition in BIO14.6 hamsters • ↑ Ca2+ reuptake in SR vesicles • ↑ Myocyte contractility and Ca2+ handling • ↑ LV systolic and diastolic function • ↓ Fibrosis • ↑ Survival PLN ablation in mdx mice • ↑ Myocyte contractility and Ca2+ handling • ↓ LV systolic and diastolic function • ↑ EBD uptake • ↑ Fibrosis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Law, M.L.; Cohen, H.; Martin, A.A.; Angulski, A.B.B.; Metzger, J.M. Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies. J. Clin. Med. 2020, 9, 520. https://doi.org/10.3390/jcm9020520
Law ML, Cohen H, Martin AA, Angulski ABB, Metzger JM. Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies. Journal of Clinical Medicine. 2020; 9(2):520. https://doi.org/10.3390/jcm9020520
Chicago/Turabian StyleLaw, Michelle L., Houda Cohen, Ashley A. Martin, Addeli Bez Batti Angulski, and Joseph M. Metzger. 2020. "Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies" Journal of Clinical Medicine 9, no. 2: 520. https://doi.org/10.3390/jcm9020520
APA StyleLaw, M. L., Cohen, H., Martin, A. A., Angulski, A. B. B., & Metzger, J. M. (2020). Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies. Journal of Clinical Medicine, 9(2), 520. https://doi.org/10.3390/jcm9020520