Sepsis-Associated Encephalopathy: From Delirium to Dementia?


{kind=link}
{kind=link}
Abstract
:1. Introduction
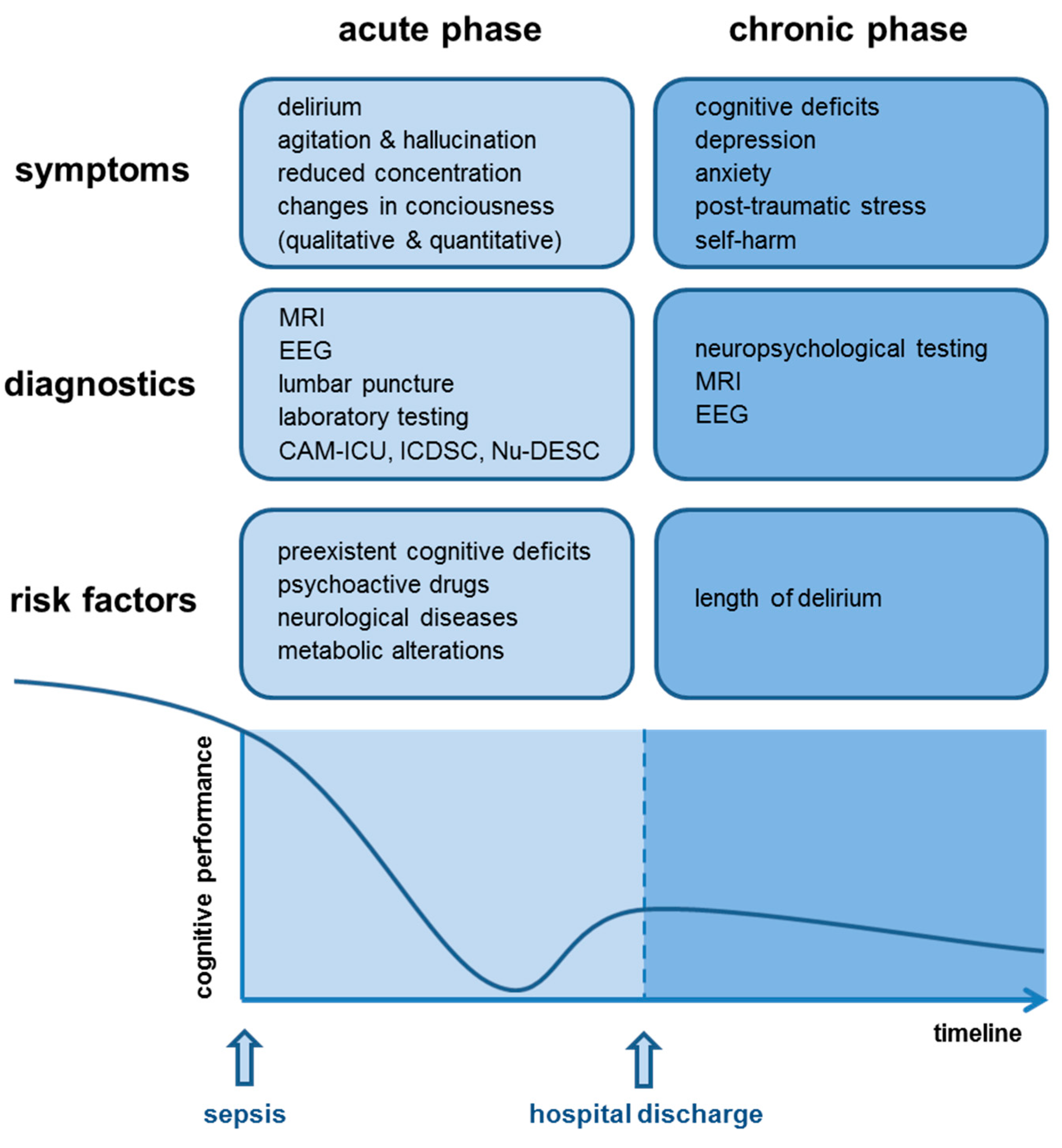
1.1. Acute Phase of SAE—Delirium
1.2. Chronic Phase of SAE—Dementia
2. Epidemiology
3. Pathophysiology
3.1. Ischemic and Hemorrhagic Cerebral Lesions
3.2. Impairment of Blood–Brain Barrier and Neuroinflammation
3.3. Dysregulated Neurotransmitter
4. Neuropathological Findings
5. Diagnostic Procedures
5.1. Cerebral Imaging
5.2. Electroencephalography
5.3. Laboratory Testing—Cerebrospinal Fluid
5.4. Laboratory Testing—Blood
5.5. Screening Tests for Delirium in Acute SAE
6. Therapeutic Management
6.1. Pharmacological Treatment
6.2. Non-Pharmacological Treatment
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K.; International Forum of Acute Care Trialists. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kempker, J.A.; Martin, G.S. The Changing Epidemiology and Definitions of Sepsis. Clin. Chest Med. 2016, 37, 165–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gofton, T.E.; Young, G.B. Sepsis-associated encephalopathy. Nat. Rev. Neurol. 2012, 8, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Heming, N.; Mazeraud, A.; Verdonk, F.; Bozza, F.A.; Chretien, F.; Sharshar, T. Neuroanatomy of sepsis-associated encephalopathy. Crit. Care 2017, 21, 65. [Google Scholar] [CrossRef] [Green Version]
- Iacobone, E.; Bailly-Salin, J.; Polito, A.; Friedman, D.; Stevens, R.D.; Sharshar, T. Sepsis-associated encephalopathy and its differential diagnosis. Crit. Care Med. 2009, 37, S331–S336. [Google Scholar] [CrossRef]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef]
- Baumgaertel, M.W.; Kraemer, M.; Berlit, P. Neurologic complications of acute and chronic renal disease. Handb. Clin. Neurol. 2014, 119, 383–393. [Google Scholar] [CrossRef]
- Sonneville, R.; de Montmollin, E.; Poujade, J.; Garrouste-Orgeas, M.; Souweine, B.; Darmon, M.; Mariotte, E.; Argaud, L.; Barbier, F.; Goldgran-Toledano, D.; et al. Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 2017, 43, 1075–1084. [Google Scholar] [CrossRef]
- Eidelman, L.A.; Putterman, D.; Putterman, C.; Sprung, C.L. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA 1996, 275, 470–473. [Google Scholar] [CrossRef]
- Pandharipande, P.P.; Girard, T.D.; Jackson, J.C.; Morandi, A.; Thompson, J.L.; Pun, B.T.; Brummel, N.E.; Hughes, C.G.; Vasilevskis, E.E.; Shintani, A.K.; et al. Long-term cognitive impairment after critical illness. N. Engl. J. Med. 2013, 369, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Girard, T.D.; Jackson, J.C.; Pandharipande, P.P.; Pun, B.T.; Thompson, J.L.; Shintani, A.K.; Gordon, S.M.; Canonico, A.E.; Dittus, R.S.; Bernard, G.R.; et al. Delirium as a predictor of long-term cognitive impairment in survivors of critical illness. Crit. Care Med. 2010, 38, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Gunther, M.L.; Morandi, A.; Krauskopf, E.; Pandharipande, P.; Girard, T.D.; Jackson, J.C.; Thompson, J.; Shintani, A.K.; Geevarghese, S.; Miller, R.R., III; et al. The association between brain volumes, delirium duration, and cognitive outcomes in intensive care unit survivors: The VISIONS cohort magnetic resonance imaging study. Crit. Care Med. 2012, 40, 2022–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, A.A.; Morrison, R.S.; Du, Q.; Wallenstein, S.; Nelson, J.E. Risk factors for long-term brain dysfunction after chronic critical illness. Ann. Am. Thorac. Soc. 2013, 10, 315–323. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annane, D.; Sharshar, T. Cognitive decline after sepsis. Lancet Respir. Med. 2015, 3, 61–69. [Google Scholar] [CrossRef]
- de Azevedo, J.R.; Montenegro, W.S.; Rodrigues, D.P.; Souza, S.C.; Araujo, V.F.; de Paula, M.P.; Prazeres, P.H.; da Luz Leitao, A.; Mendonca, A.V. Long-term cognitive outcomes among unselected ventilated and non-ventilated ICU patients. J. Intensive Care 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.C.; Girard, T.D.; Gordon, S.M.; Thompson, J.L.; Shintani, A.K.; Thomason, J.W.; Pun, B.T.; Canonico, A.E.; Dunn, J.G.; Bernard, G.R.; et al. Long-term cognitive and psychological outcomes in the awakening and breathing controlled trial. Am. J. Respir. Crit. Care Med. 2010, 182, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Needham, D.M.; Dinglas, V.D.; Morris, P.E.; Jackson, J.C.; Hough, C.L.; Mendez-Tellez, P.A.; Wozniak, A.W.; Colantuoni, E.; Ely, E.W.; Rice, T.W.; et al. Physical and cognitive performance of patients with acute lung injury 1 year after initial trophic versus full enteral feeding. EDEN trial follow-up. Am. J. Respir. Crit. Care Med. 2013, 188, 567–576. [Google Scholar] [CrossRef]
- Rothenhausler, H.B.; Ehrentraut, S.; Stoll, C.; Schelling, G.; Kapfhammer, H.P. The relationship between cognitive performance and employment and health status in long-term survivors of the acute respiratory distress syndrome: Results of an exploratory study. Gen. Hosp. Psychiatry 2001, 23, 90–96. [Google Scholar] [CrossRef]
- Sakusic, A.; O’Horo, J.C.; Dziadzko, M.; Volha, D.; Ali, R.; Singh, T.D.; Kashyap, R.; Farrell, A.M.; Fryer, J.D.; Petersen, R.; et al. Potentially Modifiable Risk Factors for Long-Term Cognitive Impairment After Critical Illness: A Systematic Review. Mayo Clin. Proc. 2018, 93, 68–82. [Google Scholar] [CrossRef] [Green Version]
- Semmler, A.; Widmann, C.N.; Okulla, T.; Urbach, H.; Kaiser, M.; Widman, G.; Mormann, F.; Weide, J.; Fliessbach, K.; Hoeft, A.; et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J. Neurol. Neurosurg. Psychiatry 2013, 84, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Prescott, H.C. Variation in Postsepsis Readmission Patterns: A Cohort Study of Veterans Affairs Beneficiaries. Ann. Am. Thorac. Soc. 2017, 14, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Iwashyna, T.J.; Speelmon, E.C. Advancing a Third Revolution in Critical Care. Am. J. Respir. Crit. Care Med. 2016, 194, 782–783. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, K.M.; Bailey, M.; Suzuki, S.; Pilcher, D.; Bellomo, R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA 2014, 311, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Wintermann, G.B.; Brunkhorst, F.M.; Petrowski, K.; Strauss, B.; Oehmichen, F.; Pohl, M.; Rosendahl, J. Stress disorders following prolonged critical illness in survivors of severe sepsis. Crit. Care Med. 2015, 43, 1213–1222. [Google Scholar] [CrossRef]
- Lund-Sorensen, H.; Benros, M.E.; Madsen, T.; Sorensen, H.J.; Eaton, W.W.; Postolache, T.T.; Nordentoft, M.; Erlangsen, A. A Nationwide Cohort Study of the Association Between Hospitalization With Infection and Risk of Death by Suicide. JAMA Psychiatry 2016, 73, 912–919. [Google Scholar] [CrossRef]
- Korosec Jagodic, H.; Jagodic, K.; Podbregar, M. Long-term outcome and quality of life of patients treated in surgical intensive care: A comparison between sepsis and trauma. Crit. Care 2006, 10, R134. [Google Scholar] [CrossRef] [Green Version]
- Langa, K.M.; Chernew, M.E.; Kabeto, M.U.; Herzog, A.R.; Ofstedal, M.B.; Willis, R.J.; Wallace, R.B.; Mucha, L.M.; Straus, W.L.; Fendrick, A.M. National estimates of the quantity and cost of informal caregiving for the elderly with dementia. J. Gen. Intern. Med. 2001, 16, 770–778. [Google Scholar] [CrossRef]
- Prescott, H.C.; Langa, K.M.; Liu, V.; Escobar, G.J.; Iwashyna, T.J. Increased 1-year healthcare use in survivors of severe sepsis. Am. J. Respir. Crit. Care Med. 2014, 190, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Angus, D.C. The lingering consequences of sepsis: A hidden public health disaster? JAMA 2010, 304, 1833–1834. [Google Scholar] [CrossRef]
- Sprung, C.L.; Peduzzi, P.N.; Shatney, C.H.; Schein, R.M.; Wilson, M.F.; Sheagren, J.N.; Hinshaw, L.B. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit. Care Med. 1990, 18, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Wang, X.T.; Ai, Y.H.; Guo, Q.L.; Huang, L.; Liu, Z.Y.; Yao, B. Epidemiological features and risk factors of sepsis-associated encephalopathy in intensive care unit patients: 2008-2011. Chin. Med. J. 2012, 125, 828–831. [Google Scholar] [PubMed]
- Sharshar, T.; Annane, D.; de la Grandmaison, G.L.; Brouland, J.P.; Hopkinson, N.S.; Francoise, G. The neuropathology of septic shock. Brain Pathol. 2004, 14, 21–33. [Google Scholar] [CrossRef]
- Polito, A.; Eischwald, F.; Maho, A.L.; Polito, A.; Azabou, E.; Annane, D.; Chretien, F.; Stevens, R.D.; Carlier, R.; Sharshar, T. Pattern of brain injury in the acute setting of human septic shock. Crit. Care 2013, 17, R204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, D.; Siegemund, M.; Dell-Kuster, S.; Smielewski, P.; Ruegg, S.; Strebel, S.P.; Marsch, S.C.; Pargger, H.; Steiner, L.A. Cerebral perfusion in sepsis-associated delirium. Crit. Care 2008, 12, R63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodson, C.M.; Rosenblatt, K.; Rivera-Lara, L.; Nyquist, P.; Hogue, C.W. Cerebral Blood Flow Autoregulation in Sepsis for the Intensivist: Why Its Monitoring May Be the Future of Individualized Care. J. Intensive Care Med. 2018, 33, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef] [Green Version]
- Crippa, I.A.; Subira, C.; Vincent, J.L.; Fernandez, R.F.; Hernandez, S.C.; Cavicchi, F.Z.; Creteur, J.; Taccone, F.S. Impaired cerebral autoregulation is associated with brain dysfunction in patients with sepsis. Crit. Care 2018, 22, 327. [Google Scholar] [CrossRef] [Green Version]
- Taccone, F.S.; Su, F.; De Deyne, C.; Abdellhai, A.; Pierrakos, C.; He, X.; Donadello, K.; Dewitte, O.; Vincent, J.L.; De Backer, D. Sepsis is associated with altered cerebral microcirculation and tissue hypoxia in experimental peritonitis. Crit. Care Med. 2014, 42, e114–e122. [Google Scholar] [CrossRef]
- Sharshar, T.; Polito, A.; Checinski, A.; Stevens, R.D. Septic-associated encephalopathy—Everything starts at a microlevel. Crit. Care 2010, 14, 199. [Google Scholar] [CrossRef] [Green Version]
- Nwafor, D.C.; Brichacek, A.L.; Mohammad, A.S.; Griffith, J.; Lucke-Wold, B.P.; Benkovic, S.A.; Geldenhuys, W.J.; Lockman, P.R.; Brown, C.M. Targeting the Blood-Brain Barrier to Prevent Sepsis-Associated Cognitive Impairment. J. Cent. Nerv. Syst. Dis. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Handa, O.; Stephen, J.; Cepinskas, G. Role of endothelial nitric oxide synthase-derived nitric oxide in activation and dysfunction of cerebrovascular endothelial cells during early onsets of sepsis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1712–H1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuperberg, S.J.; Wadgaonkar, R. Sepsis-Associated Encephalopathy: The Blood-Brain Barrier and the Sphingolipid Rheostat. Front. Immunol. 2017, 8, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulyatnikova, T.; Verkhratsky, A. Astroglia in Sepsis Associated Encephalopathy. Neurochem. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Singh, S.K.; Stogsdill, J.A.; Pulimood, N.S.; Dingsdale, H.; Kim, Y.H.; Pilaz, L.J.; Kim, I.H.; Manhaes, A.C.; Rodrigues, W.S., Jr.; Pamukcu, A.; et al. Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1alpha and NL1 via Hevin. Cell 2016, 164, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.V.; Bernardinelli, Y.; Tse, Y.C.; Chierzi, S.; Wong, T.P.; Murai, K.K. Astrocytes control glutamate receptor levels at developing synapses through SPARC-beta-integrin interactions. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 4154–4165. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, M.; Sonai, B.; Dal-Pizzol, F. Polarization of microglia and its role in bacterial sepsis. J. Neuroimmunol. 2017, 303, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Lemstra, A.W.; Groen in’t Woud, J.C.; Hoozemans, J.J.; van Haastert, E.S.; Rozemuller, A.J.; Eikelenboom, P.; van Gool, W.A. Microglia activation in sepsis: A case-control study. J. Neuroinflammation 2007, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Guo, C.; Hou, W.; Shen, N.; Miao, C. Effects of MFHAS1 on cognitive impairment and dendritic pathology in the hippocampus of septic rats. Life Sci. 2019, 235, 116822. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, A.; Lerman, Y.V.; Kim, T.H.; Kim, M.R.; Mai, N.; Halterman, M.W.; Kim, M. Long-Term Microgliosis Driven by Acute Systemic Inflammation. J. Immunol. 2019, 203, 2979–2989. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Zelinski, E.L.; Schubert, C.L.; Knight, D.; Craig, L.A.; Winston, B.W.; Spanswick, S.C.; Petri, B.; Jenne, C.N.; Sutherland, J.C.; et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- van Gool, W.A.; van de Beek, D.; Eikelenboom, P. Systemic infection and delirium: When cytokines and acetylcholine collide. Lancet 2010, 375, 773–775. [Google Scholar] [CrossRef]
- Semmler, A.; Frisch, C.; Debeir, T.; Ramanathan, M.; Okulla, T.; Klockgether, T.; Heneka, M.T. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp. Neurol. 2007, 204, 733–740. [Google Scholar] [CrossRef]
- Zhai, Q.; Lai, D.; Cui, P.; Zhou, R.; Chen, Q.; Hou, J.; Su, Y.; Pan, L.; Ye, H.; Zhao, J.W.; et al. Selective Activation of Basal Forebrain Cholinergic Neurons Attenuates Polymicrobial Sepsis-Induced Inflammation via the Cholinergic Anti-Inflammatory Pathway. Crit. Care Med. 2017, 45, e1075–e1082. [Google Scholar] [CrossRef] [Green Version]
- Osuchowski, M.F.; Ayala, A.; Bahrami, S.; Bauer, M.; Boros, M.; Cavaillon, J.M.; Chaudry, I.H.; Coopersmith, C.M.; Deutschman, C.S.; Drechsler, S.; et al. Minimum Quality Threshold in Pre-Clinical Sepsis Studies (MQTiPSS): An International Expert Consensus Initiative for Improvement of Animal Modeling in Sepsis. Shock 2018, 50, 377–380. [Google Scholar] [CrossRef] [Green Version]
- van Eijk, M.M.; Roes, K.C.; Honing, M.L.; Kuiper, M.A.; Karakus, A.; van der Jagt, M.; Spronk, P.E.; van Gool, W.A.; van der Mast, R.C.; Kesecioglu, J.; et al. Effect of rivastigmine as an adjunct to usual care with haloperidol on duration of delirium and mortality in critically ill patients: A multicentre, double-blind, placebo-controlled randomised trial. Lancet 2010, 376, 1829–1837. [Google Scholar] [CrossRef] [Green Version]
- Basler, T.; Meier-Hellmann, A.; Bredle, D.; Reinhart, K. Amino acid imbalance early in septic encephalopathy. Intensive Care Med. 2002, 28, 293–298. [Google Scholar] [CrossRef]
- Jackson, A.C.; Gilbert, J.J.; Young, G.B.; Bolton, C.F. The encephalopathy of sepsis. Can. J. Neurol. Sci. 1985, 12, 303–307. [Google Scholar] [CrossRef] [Green Version]
- Sharshar, T.; Gray, F.; Lorin de la Grandmaison, G.; Hopkinson, N.S.; Ross, E.; Dorandeu, A.; Orlikowski, D.; Raphael, J.C.; Gajdos, P.; Annane, D. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 2003, 362, 1799–1805. [Google Scholar] [CrossRef]
- Sharshar, T.; Gray, F.; Poron, F.; Raphael, J.C.; Gajdos, P.; Annane, D. Multifocal necrotizing leukoencephalopathy in septic shock. Crit. Care Med. 2002, 30, 2371–2375. [Google Scholar] [CrossRef]
- Westhoff, D.; Engelen-Lee, J.Y.; Hoogland, I.C.M.; Aronica, E.M.A.; van Westerloo, D.J.; van de Beek, D.; van Gool, W.A. Systemic infection and microglia activation: A prospective postmortem study in sepsis patients. Immun. Ageing I A 2019, 16, 18. [Google Scholar] [CrossRef] [Green Version]
- Zrzavy, T.; Hoftberger, R.; Berger, T.; Rauschka, H.; Butovsky, O.; Weiner, H.; Lassmann, H. Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropathol. Appl. Neurobiol. 2019, 45, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, D.J.; Yamamoto, A.K.; Menon, D.K. Imaging in sepsis-associated encephalopathy—Insights and opportunities. Nat. Rev. Neurol. 2013, 9, 551–561. [Google Scholar] [CrossRef]
- Bartynski, W.S.; Boardman, J.F.; Zeigler, Z.R.; Shadduck, R.K.; Lister, J. Posterior reversible encephalopathy syndrome in infection, sepsis, and shock. AJNR Am. J. Neuroradiol. 2006, 27, 2179–2190. [Google Scholar]
- Young, G.B.; Bolton, C.F.; Archibald, Y.M.; Austin, T.W.; Wells, G.A. The electroencephalogram in sepsis-associated encephalopathy. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 1992, 9, 145–152. [Google Scholar] [CrossRef]
- Hosokawa, K.; Gaspard, N.; Su, F.; Oddo, M.; Vincent, J.L.; Taccone, F.S. Clinical neurophysiological assessment of sepsis-associated brain dysfunction: A systematic review. Crit. Care 2014, 18, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azabou, E.; Magalhaes, E.; Braconnier, A.; Yahiaoui, L.; Moneger, G.; Heming, N.; Annane, D.; Mantz, J.; Chretien, F.; Durand, M.C.; et al. Early Standard Electroencephalogram Abnormalities Predict Mortality in Septic Intensive Care Unit Patients. PLoS ONE 2015, 10, e0139969. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.M.; Urdanibia-Centelles, O.; Vedel-Larsen, E.; Thomsen, K.J.; Moller, K.; Olsen, K.S.; Lauritsen, A.O.; Eddelien, H.S.; Lauritzen, M.; Benedek, K. Continuous EEG Monitoring in a Consecutive Patient Cohort with Sepsis and Delirium. Neurocrit. Care 2019. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, P.; Gaspard, N.; Wahl, A.S.; Bauer, R.M.; Hirsch, L.J.; Wunsch, H.; Claassen, J. Continuous electroencephalography in a surgical intensive care unit. Intensive Care Med. 2014, 40, 228–234. [Google Scholar] [CrossRef]
- Nguyen, D.N.; Spapen, H.; Su, F.; Schiettecatte, J.; Shi, L.; Hachimi-Idrissi, S.; Huyghens, L. Elevated serum levels of S-100beta protein and neuron-specific enolase are associated with brain injury in patients with severe sepsis and septic shock. Crit. Care Med. 2006, 34, 1967–1974. [Google Scholar] [CrossRef]
- Anderson, B.J.; Reilly, J.P.; Shashaty, M.G.S.; Palakshappa, J.A.; Wysoczanski, A.; Dunn, T.G.; Kazi, A.; Tommasini, A.; Mikkelsen, M.E.; Schweickert, W.D.; et al. Admission plasma levels of the neuronal injury marker neuron-specific enolase are associated with mortality and delirium in sepsis. J. Crit. Care 2016, 36, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Ehler, J.; Petzold, A.; Wittstock, M.; Kolbaske, S.; Gloger, M.; Henschel, J.; Heslegrave, A.; Zetterberg, H.; Lunn, M.P.; Rommer, P.S.; et al. The prognostic value of neurofilament levels in patients with sepsis-associated encephalopathy—A prospective, pilot observational study. PLoS ONE 2019, 14, e0211184. [Google Scholar] [CrossRef] [Green Version]
- Shahim, P.; Zetterberg, H.; Tegner, Y.; Blennow, K. Serum neurofilament light as a biomarker for mild traumatic brain injury in contact sports. Neurology 2017, 88, 1788–1794. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.H.; Macdonald-Wallis, C.; Gray, E.; Pearce, N.; Petzold, A.; Norgren, N.; Giovannoni, G.; Fratta, P.; Sidle, K.; Fish, M.; et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015, 84, 2247–2257. [Google Scholar] [CrossRef] [Green Version]
- Kuhle, J.; Nourbakhsh, B.; Grant, D.; Morant, S.; Barro, C.; Yaldizli, O.; Pelletier, D.; Giovannoni, G.; Waubant, E.; Gnanapavan, S. Serum neurofilament is associated with progression of brain atrophy and disability in early MS. Neurology 2017, 88, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Ely, E.W.; Margolin, R.; Francis, J.; May, L.; Truman, B.; Dittus, R.; Speroff, T.; Gautam, S.; Bernard, G.R.; Inouye, S.K. Evaluation of delirium in critically ill patients: Validation of the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU). Crit. Care Med. 2001, 29, 1370–1379. [Google Scholar] [CrossRef]
- Bergeron, N.; Dubois, M.J.; Dumont, M.; Dial, S.; Skrobik, Y. Intensive Care Delirium Screening Checklist: Evaluation of a new screening tool. Intensive Care Med. 2001, 27, 859–864. [Google Scholar] [CrossRef]
- Luetz, A.; Heymann, A.; Radtke, F.M.; Chenitir, C.; Neuhaus, U.; Nachtigall, I.; von Dossow, V.; Marz, S.; Eggers, V.; Heinz, A.; et al. Different assessment tools for intensive care unit delirium: Which score to use? Crit. Care Med. 2010, 38, 409–418. [Google Scholar] [CrossRef]
- Inouye, S.K.; van Dyck, C.H.; Alessi, C.A.; Balkin, S.; Siegal, A.P.; Horwitz, R.I. Clarifying confusion: The confusion assessment method. A new method for detection of delirium. Ann. Intern. Med. 1990, 113, 941–948. [Google Scholar] [CrossRef]
- Marcantonio, E.R.; Ngo, L.H.; O’Connor, M.; Jones, R.N.; Crane, P.K.; Metzger, E.D.; Inouye, S.K. 3D-CAM: Derivation and validation of a 3-minute diagnostic interview for CAM-defined delirium: A cross-sectional diagnostic test study. Ann. Intern. Med. 2014, 161, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Inouye, S.K.; Kosar, C.M.; Tommet, D.; Schmitt, E.M.; Puelle, M.R.; Saczynski, J.S.; Marcantonio, E.R.; Jones, R.N. The CAM-S: Development and validation of a new scoring system for delirium severity in 2 cohorts. Ann. Intern. Med. 2014, 160, 526–533. [Google Scholar] [CrossRef] [Green Version]
- Gaudreau, J.D.; Gagnon, P.; Harel, F.; Tremblay, A.; Roy, M.A. Fast, systematic, and continuous delirium assessment in hospitalized patients: The nursing delirium screening scale. J. Pain Symptom Manag. 2005, 29, 368–375. [Google Scholar] [CrossRef]
- Bellelli, G.; Morandi, A.; Davis, D.H.; Mazzola, P.; Turco, R.; Gentile, S.; Ryan, T.; Cash, H.; Guerini, F.; Torpilliesi, T.; et al. Validation of the 4AT, a new instrument for rapid delirium screening: A study in 234 hospitalised older people. Age Ageing 2014, 43, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Luetz, A.; Balzer, F.; Radtke, F.M.; Jones, C.; Citerio, G.; Walder, B.; Weiss, B.; Wernecke, K.D.; Spies, C. Delirium, sedation and analgesia in the intensive care unit: A multinational, two-part survey among intensivists. PLoS ONE 2014, 9, e110935. [Google Scholar] [CrossRef] [Green Version]
- Barr, J.; Fraser, G.L.; Puntillo, K.; Ely, E.W.; Gelinas, C.; Dasta, J.F.; Davidson, J.E.; Devlin, J.W.; Kress, J.P.; Joffe, A.M.; et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit. Care Med. 2013, 41, 263–306. [Google Scholar] [CrossRef]
- Girard, T.D.; Thompson, J.L.; Pandharipande, P.P.; Brummel, N.E.; Jackson, J.C.; Patel, M.B.; Hughes, C.G.; Chandrasekhar, R.; Pun, B.T.; Boehm, L.M.; et al. Clinical phenotypes of delirium during critical illness and severity of subsequent long-term cognitive impairment: A prospective cohort study. Lance Respir. Med. 2018, 6, 213–222. [Google Scholar] [CrossRef]
- Girard, T.D.; Exline, M.C.; Carson, S.S.; Hough, C.L.; Rock, P.; Gong, M.N.; Douglas, I.S.; Malhotra, A.; Owens, R.L.; Feinstein, D.J.; et al. Haloperidol and Ziprasidone for Treatment of Delirium in Critical Illness. N. Engl. J. Med. 2018, 379, 2506–2516. [Google Scholar] [CrossRef]
- Nikooie, R.; Neufeld, K.J.; Oh, E.S.; Wilson, L.M.; Zhang, A.; Robinson, K.A.; Needham, D.M. Antipsychotics for Treating Delirium in Hospitalized Adults: A Systematic Review. Ann. Intern. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Campbell, N.; Boustani, M.; Limbil, T.; Ott, C.; Fox, C.; Maidment, I.; Schubert, C.C.; Munger, S.; Fick, D.; Miller, D.; et al. The cognitive impact of anticholinergics: A clinical review. Clin. Interv. Aging 2009, 4, 225–233. [Google Scholar]
- Pandharipande, P.P.; Sanders, R.D.; Girard, T.D.; McGrane, S.; Thompson, J.L.; Shintani, A.K.; Herr, D.L.; Maze, M.; Ely, E.W. Effect of dexmedetomidine versus lorazepam on outcome in patients with sepsis: An a priori-designed analysis of the MENDS randomized controlled trial. Crit. Care 2010, 14, R38. [Google Scholar] [CrossRef] [Green Version]
- Kawazoe, Y.; Miyamoto, K.; Morimoto, T.; Yamamoto, T.; Fuke, A.; Hashimoto, A.; Koami, H.; Beppu, S.; Katayama, Y.; Itoh, M.; et al. Effect of Dexmedetomidine on Mortality and Ventilator-Free Days in Patients Requiring Mechanical Ventilation With Sepsis: A Randomized Clinical Trial. JAMA 2017, 317, 1321–1328. [Google Scholar] [CrossRef]
- Morandi, A.; Hughes, C.G.; Thompson, J.L.; Pandharipande, P.P.; Shintani, A.K.; Vasilevskis, E.E.; Han, J.H.; Jackson, J.C.; Laskowitz, D.T.; Bernard, G.R.; et al. Statins and delirium during critical illness: A multicenter, prospective cohort study. Crit. Care Med. 2014, 42, 1899–1909. [Google Scholar] [CrossRef]
- Lee, C.C.; Lee, M.G.; Hsu, T.C.; Porta, L.; Chang, S.S.; Yo, C.H.; Tsai, K.C.; Lee, M. A Population-Based Cohort Study on the Drug-Specific Effect of Statins on Sepsis Outcome. Chest 2018, 153, 805–815. [Google Scholar] [CrossRef]
- Schweickert, W.D.; Pohlman, M.C.; Pohlman, A.S.; Nigos, C.; Pawlik, A.J.; Esbrook, C.L.; Spears, L.; Miller, M.; Franczyk, M.; Deprizio, D.; et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: A randomised controlled trial. Lancet 2009, 373, 1874–1882. [Google Scholar] [CrossRef]
- Brummel, N.E.; Girard, T.D.; Ely, E.W.; Pandharipande, P.P.; Morandi, A.; Hughes, C.G.; Graves, A.J.; Shintani, A.; Murphy, E.; Work, B.; et al. Feasibility and safety of early combined cognitive and physical therapy for critically ill medical and surgical patients: The Activity and Cognitive Therapy in ICU (ACT-ICU) trial. Intensive Care Med. 2014, 40, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Ke, L.; Wang, J.; Ma, Z.; Chen, B.; Wang, L.; Gong, J.; Wang, R. Non-pharmacological Treatment of Intensive Care Unit Delirium. Am. J. Nurs. Sci. 2019, 8, 119–128. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, H.-Y.; Wickel, J.; Brunkhorst, F.M.; Geis, C. Sepsis-Associated Encephalopathy: From Delirium to Dementia? J. Clin. Med. 2020, 9, 703. https://doi.org/10.3390/jcm9030703
Chung H-Y, Wickel J, Brunkhorst FM, Geis C. Sepsis-Associated Encephalopathy: From Delirium to Dementia? Journal of Clinical Medicine. 2020; 9(3):703. https://doi.org/10.3390/jcm9030703
Chicago/Turabian StyleChung, Ha-Yeun, Jonathan Wickel, Frank M. Brunkhorst, and Christian Geis. 2020. "Sepsis-Associated Encephalopathy: From Delirium to Dementia?" Journal of Clinical Medicine 9, no. 3: 703. https://doi.org/10.3390/jcm9030703
APA StyleChung, H.-Y., Wickel, J., Brunkhorst, F. M., & Geis, C. (2020). Sepsis-Associated Encephalopathy: From Delirium to Dementia? Journal of Clinical Medicine, 9(3), 703. https://doi.org/10.3390/jcm9030703