Identification of Candidate Genes Associated with Susceptibility to Ovarian Clear Cell Adenocarcinoma Using cis-eQTL Analysis


 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Susceptibility Loci and Associated Genes through cis-eQTL Analysis
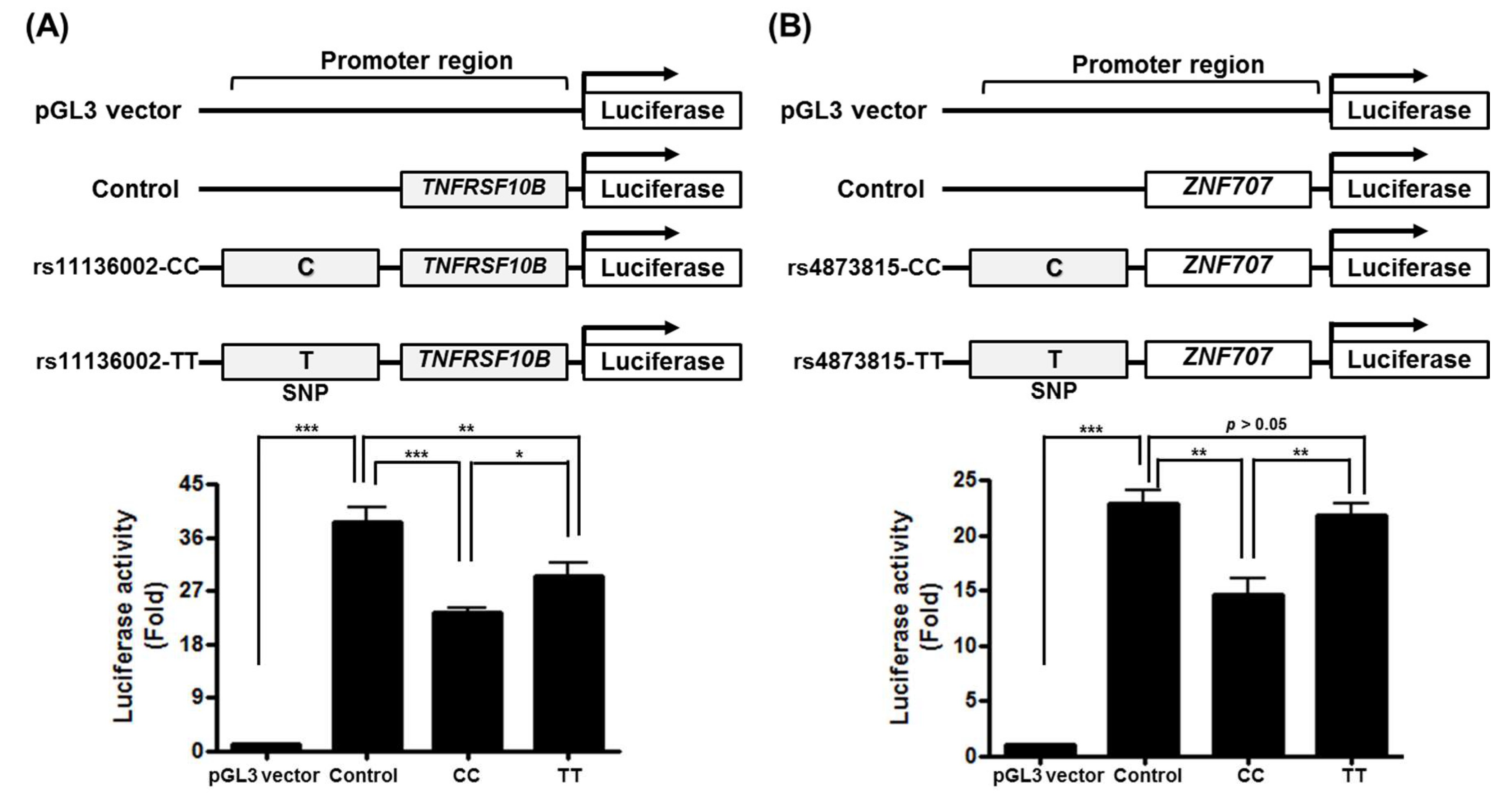
2.2. Functional Validation of Susceptibility Loci on Associated Genes
2.3. Analysis of the Clinical Significance of the Seven Identified Genes Using Our Sample Cohort
2.4. Analysis of the Clinical Significance of the Seven Identified Genes Using TCGA Datasets
2.5. Statistical Analysis
3. Results
3.1. Total of 935 SNPs Identified through cis-eQTL Analysis of 94 Genes Reported in Ov-CCA
3.2. Four cis-eQTL with Differential Allelic Frequency in the Japanese Ancestry
3.3. Seven Genes Associated with Four SNPs in cis
3.4. Potential Regulatory Effect of rs4873815-TT and rs11136002-TT on Associate Genes
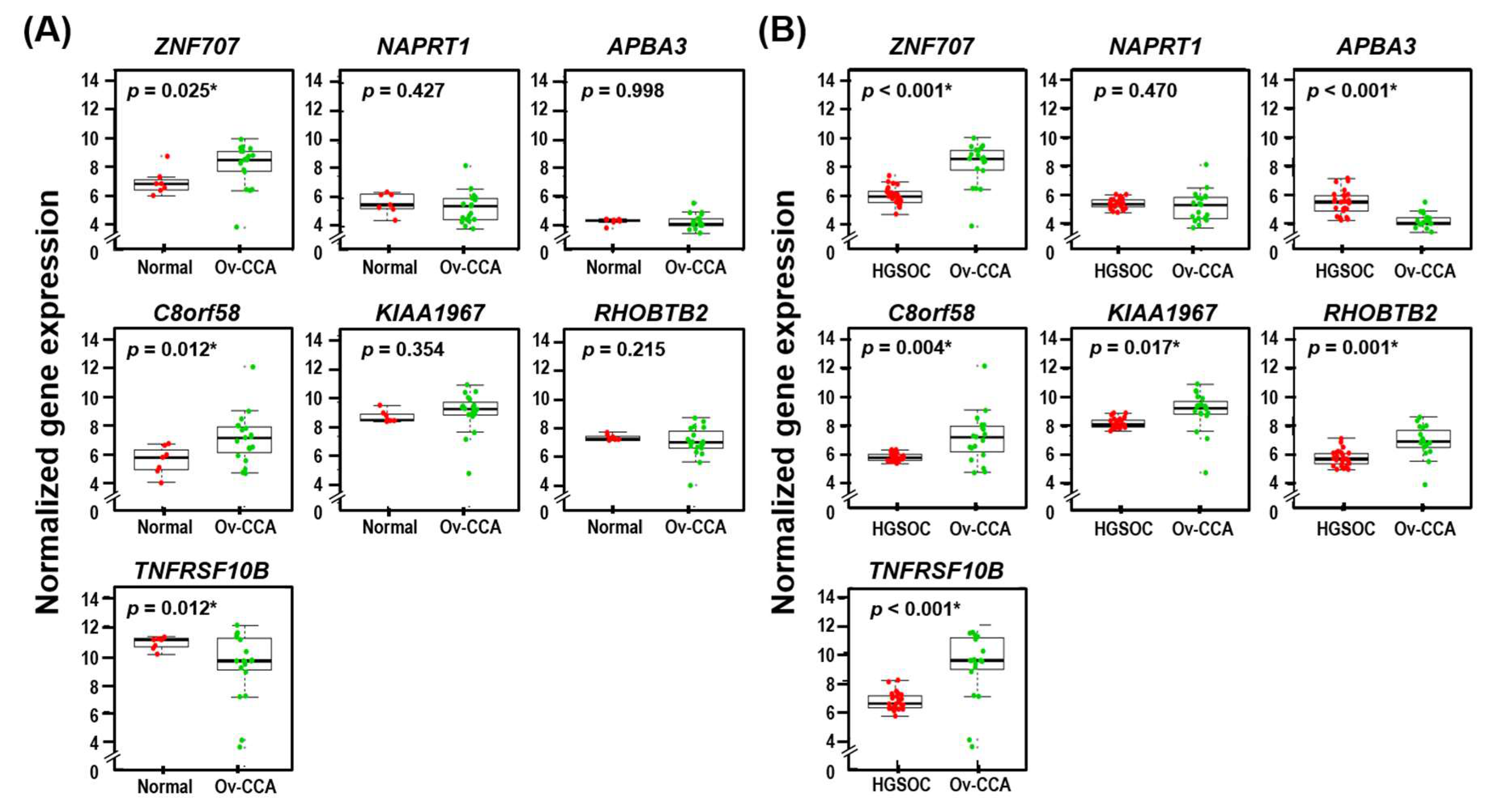
3.5. Seven Identified Genes Were Differentially Expressed in Ov-CCA Compared to Normal or HGSOC
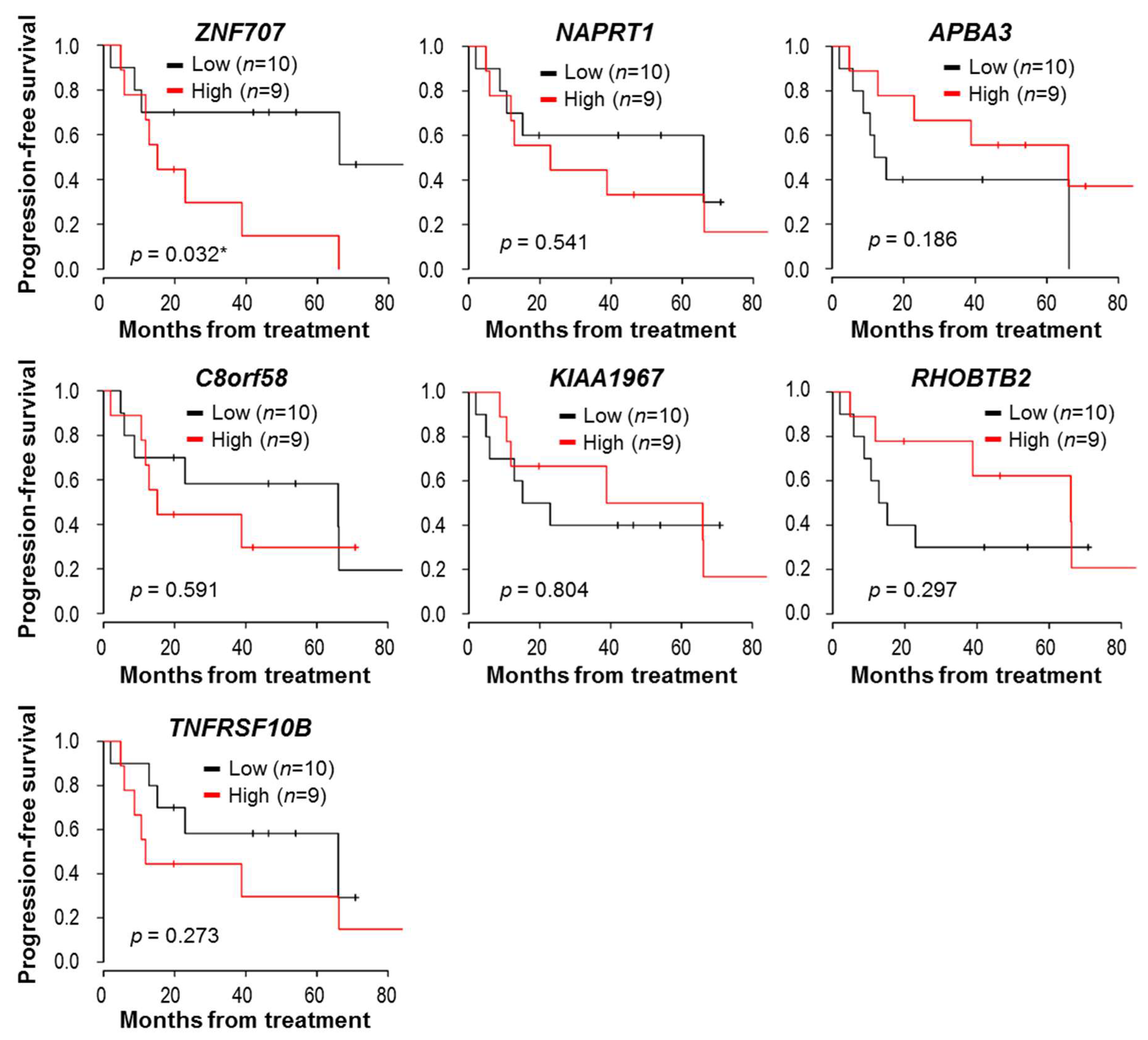
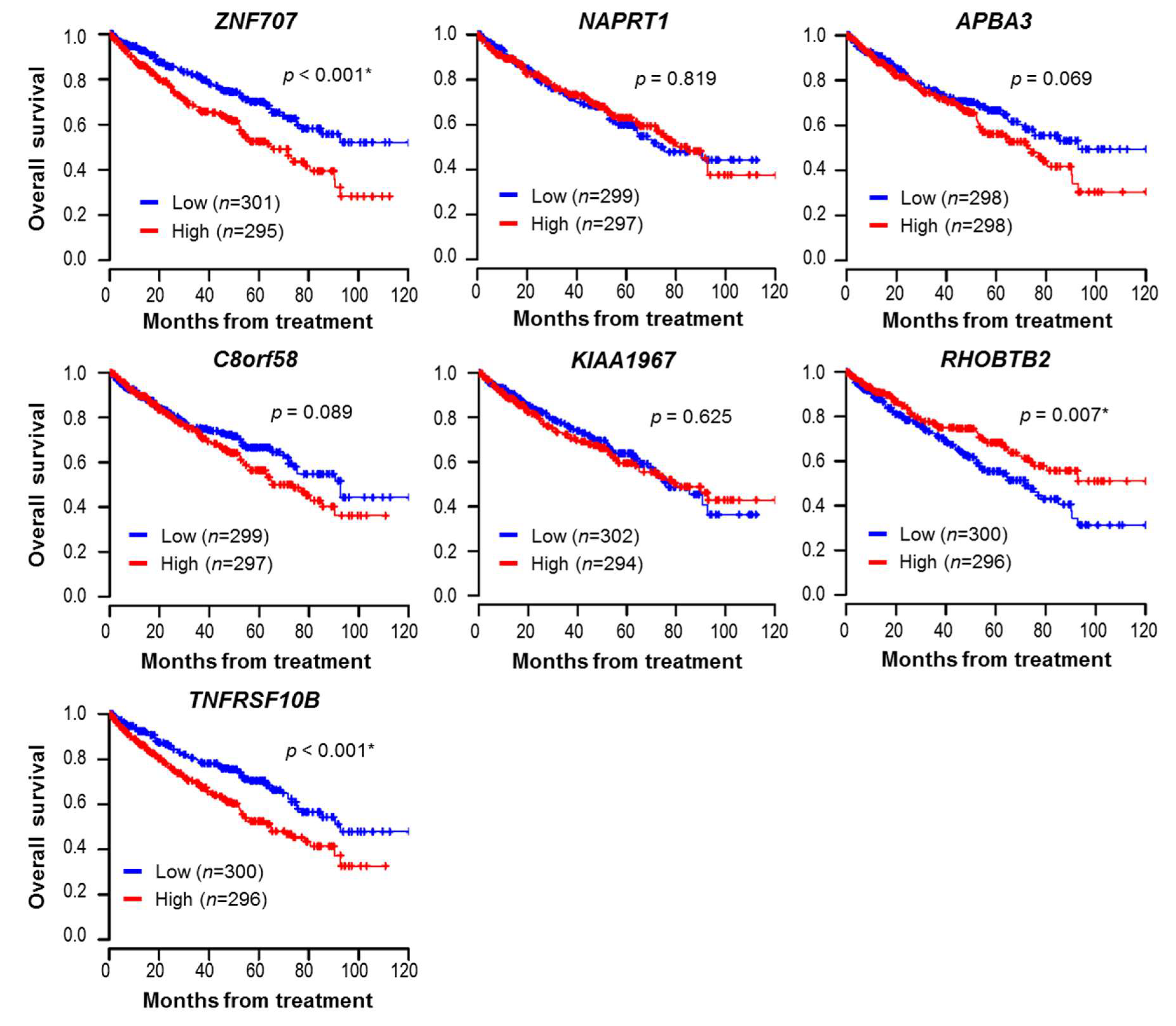
3.6. Prognostic Significance of the Identified Genes in Clear Cell Histology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120362. [Google Scholar] [CrossRef]
- Chen, Q.R.; Hu, Y.; Yan, C.; Buetow, K.; Meerzaman, D. Systematic genetic analysis identifies Cis-eQTL target genes associated with glioblastoma patient survival. PLoS ONE 2014, 9, e105393. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Stram, A.; Chen, C.; Kar, S.; Gayther, S.; Pharoah, P.; Haiman, C.; Stranger, B.; Kraft, P.; Freedman, M.L. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Hum. Mol. Genet. 2014, 23, 5294–5302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolio, T.A. Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 2010, 363, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, M.L.; Monteiro, A.N.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; De Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 2011, 43, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Kelemen, L.E.; Lawrenson, K.; Tyrer, J.; Li, Q.; Lee, J.M.; Seo, J.H.; Phelan, C.M.; Beesley, J.; Chen, X.; Spindler, T.J.; et al. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat. Genet. 2015, 47, 888–897. [Google Scholar] [CrossRef]
- Lawrenson, K.; Song, F.; Hazelett, D.J.; Kar, S.P.; Tyrer, J.; Phelan, C.M.; Corona, R.I.; Rodriguez-Malave, N.I.; Seo, J.H.; Adler, E.; et al. Genome-wide association studies identify susceptibility loci for epithelial ovarian cancer in east Asian women. Gynecol. Oncol. 2019, 153, 343–355. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Shrestha, Y.; Flavin, R.J.; Regan, M.M.; Penney, K.L.; Mucci, L.A.; Stampfer, M.J.; Hunter, D.J.; Chanock, S.J.; Schafer, E.J.; et al. Analysis of the 10q11 cancer risk locus implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet. 2010, 6, e1001204. [Google Scholar] [CrossRef]
- Li, Q.; Seo, J.H.; Stranger, B.; McKenna, A.; Pe’er, I.; Laframboise, T.; Brown, M.; Tyekucheva, S.; Freedman, M.L. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013, 152, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Lawrenson, K.; Li, Q.; Kar, S.; Seo, J.H.; Tyrer, J.; Spindler, T.J.; Lee, J.; Chen, Y.; Karst, A.; Drapkin, R.; et al. Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat. Commun. 2015, 6, 8234. [Google Scholar] [CrossRef] [PubMed]
- McGuire, V.; Jesser, C.A.; Whittemore, A.S. Survival among U.S. women with invasive epithelial ovarian cancer. Gynecol. Oncol. 2002, 84, 399–403. [Google Scholar] [CrossRef]
- Heintz, A.P.; Odicino, F.; Maisonneuve, P.; Quinn, M.A.; Benedet, J.L.; Creasman, W.T.; Ngan, H.Y.; Pecorelli, S.; Beller, U. Carcinoma of the ovary. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int. J. Gynaecol. Obstet. 2006, 95 (Suppl. 1), S161–S192. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Carey, M.S.; Kobel, M.; Mackay, H.; Huntsman, D.G. Vancouver Ovarian Clear Cell Symposium, S. Clear cell carcinoma of the ovary: A report from the first Ovarian Clear Cell Symposium, June 24th, 2010. Gynecol. Oncol. 2011, 121, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Recent concepts of ovarian carcinogenesis: Type I and type II. Biomed. Res. Int. 2014, 2014, 934261. [Google Scholar] [CrossRef] [Green Version]
- Fejerman, L.; Stern, M.C.; John, E.M.; Torres-Mejia, G.; Hines, L.M.; Wolff, R.K.; Baumgartner, K.B.; Giuliano, A.R.; Ziv, E.; Perez-Stable, E.J.; et al. Interaction between common breast cancer susceptibility variants, genetic ancestry, and nongenetic risk factors in Hispanic women. Cancer Epidemiol. Prev. Biomark. 2015, 24, 1731–1738. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.; Yuan, J.; Kensler, K.H.; Hu, Z.; Zhang, Y.; Zhang, T.; Jiang, J.; Xu, M.; Pan, Y.; Long, M.; et al. Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry. PLoS Genet. 2020, 16. [Google Scholar] [CrossRef] [Green Version]
- Sung, C.O.; Choi, C.H.; Ko, Y.H.; Ju, H.; Choi, Y.L.; Kim, N.; Kang, S.Y.; Ha, S.Y.; Choi, K.; Bae, D.S.; et al. Integrative analysis of copy number alteration and gene expression profiling in ovarian clear cell adenocarcinoma. Cancer Genet. 2013, 206, 145–153. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.P.; Beazley, C.; Montgomery, S.B.; Dimas, A.S.; Gutierrez-Arcelus, M.; Stranger, B.E.; Deloukas, P.; Dermitzakis, E.T. Genevar: A database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics 2010, 26, 2474–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadista, J.; Oskolkov, N.; Hansson, O.; Groop, L. LoFtool: A gene intolerance score based on loss-of-function variants in 60 706 individuals. Bioinformatics 2017, 33, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; de Ridder, D.; Reinders, M. Predicting variant deleteriousness in non-human species: Applying the CADD approach in mouse. BMC Bioinform. 2018, 19, 373. [Google Scholar] [CrossRef]
- Liu, L.; Pei, Y.F.; Liu, T.L.; Hu, W.Z.; Yang, X.L.; Li, S.C.; Hai, R.; Ran, S.; Zhao, L.J.; Shen, H.; et al. Identification of a 1p21 independent functional variant for abdominal obesity. Int. J. Obes. (London) 2019, 43, 2480–2490. [Google Scholar] [CrossRef]
- Choi, C.H.; Choi, J.J.; Park, Y.A.; Lee, Y.Y.; Song, S.Y.; Sung, C.O.; Song, T.; Kim, M.K.; Kim, T.J.; Lee, J.W.; et al. Identification of differentially expressed genes according to chemosensitivity in advanced ovarian serous adenocarcinomas: Expression of GRIA2 predicts better survival. Br. J. Cancer 2012, 107, 91–99. [Google Scholar] [CrossRef]
- Taminau, J.; Meganck, S.; Lazar, C.; Steenhoff, D.; Coletta, A.; Molter, C.; Duque, R.; de Schaetzen, V.; Weiss Solis, D.Y.; Bersini, H.; et al. Unlocking the potential of publicly available microarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinform. 2012, 13, 335. [Google Scholar] [CrossRef]
- Ji, J.X.; Wang, Y.K.; Cochrane, D.R.; Huntsman, D.G. Clear cell carcinomas of the ovary and kidney: Clarity through genomics. J. Pathol. 2018, 244, 550–564. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Lee, Y.S.; Kim, J.; Chung, J.Y.; Kim, J.H. Overexpression of glucose transporter-1 (GLUT-1) predicts poor prognosis in epithelial ovarian cancer. Cancer Investig. 2013, 31, 607–615. [Google Scholar] [CrossRef]
- Borghese, B.; Barbaux, S.; Mondon, F.; Santulli, P.; Pierre, G.; Vinci, G.; Chapron, C.; Vaiman, D. Research resource: Genome-wide profiling of methylated promoters in endometriosis reveals a subtelomeric location of hypermethylation. Mol. Endocrinol. 2010, 24, 1872–1885. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Shin, M.S.; Kim, H.S.; Lee, H.K.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Han, S.Y.; Park, J.Y.; Oh, R.R.; et al. Somatic mutations of TRAIL-receptor 1 and TRAIL-receptor 2 genes in non-Hodgkin’s lymphoma. Oncogene 2001, 20, 399–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsenlehner, T.; Langsenlehner, U.; Renner, W.; Kapp, K.S.; Krippl, P.; Hofmann, G.; Clar, H.; Pummer, K.; Mayer, R. The Glu228Ala polymorphism in the ligand binding domain of death receptor 4 is associated with increased risk for prostate cancer metastases. Prostate 2008, 68, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Chae, Y.S.; Moon, J.H.; Kang, B.W.; Kim, J.G.; Sohn, S.K.; Park, J.Y.; Lee, M.H.; Park, H.Y. TNF superfamily gene polymorphism as prognostic factor in early breast cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Song, C.; Liu, J.; Chen, J.; Zhai, X.; Hu, Z. Expression quantitative trait loci for TNFRSF10 influence both HBV infection and hepatocellular carcinoma development. J. Med. Virol. 2016, 88, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, X.; Su, L. Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, U.; Kronung, S.K.; Leha, A.; Walter, L.; Dobbelstein, M. Comprehensive identification of genes driven by ERV9-LTRs reveals TNFRSF10B as a re-activatable mediator of testicular cancer cell death. Cell Death Differ. 2016, 23, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emi, M.; Fujiwara, Y.; Nakajima, T.; Tsuchiya, E.; Tsuda, H.; Hirohashi, S.; Maeda, Y.; Tsuruta, K.; Miyaki, M.; Nakamura, Y. Frequent loss of heterozygosity for loci on chromosome 8p in hepatocellular carcinoma, colorectal cancer, and lung cancer. Cancer Res. 1992, 52, 5368–5372. [Google Scholar]
- Lundgren, R.; Mandahl, N.; Heim, S.; Limon, J.; Henrikson, H.; Mitelman, F. Cytogenetic analysis of 57 primary prostatic adenocarcinomas. Genes Chromosomes Cancer 1992, 4, 16–24. [Google Scholar] [CrossRef]
- Knowles, M.A.; Aveyard, J.S.; Taylor, C.F.; Harnden, P.; Bass, S. Mutation analysis of the 8p candidate tumour suppressor genes DBC2 (RHOBTB2) and LZTS1 in bladder cancer. Cancer Lett. 2005, 225, 121–130. [Google Scholar] [CrossRef]
- Mao, H.; Qu, X.; Yang, Y.; Zuo, W.; Bi, Y.; Zhou, C.; Yin, H.; Deng, B.; Sun, J.; Zhang, L. A novel tumor suppressor gene RhoBTB2 (DBC2): Frequent loss of expression in sporadic breast cancer. Mol. Carcinog. 2010, 49, 283–289. [Google Scholar] [CrossRef]
- Hara, T.; Nakaoka, H.J.; Hayashi, T.; Mimura, K.; Hoshino, D.; Inoue, M.; Nagamura, F.; Murakami, Y.; Seiki, M.; Sakamoto, T. Control of metastatic niche formation by targeting APBA3/Mint3 in inflammatory monocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E4416–E4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, X.; Zhang, X.; Chen, R. Identification of important long non-coding RNAs and highly recurrent aberrant alternative splicing events in hepatocellular carcinoma through integrative analysis of multiple RNA-Seq datasets. Mol. Genet. Genom. 2016, 291, 1035–1051. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piacente, F.; Caffa, I.; Ravera, S.; Sociali, G.; Passalacqua, M.; Vellone, V.G.; Becherini, P.; Reverberi, D.; Monacelli, F.; Ballestrero, A.; et al. Nicotinic Acid Phosphoribosyltransferase Regulates Cancer Cell Metabolism, Susceptibility to NAMPT Inhibitors, and DNA Repair. Cancer Res. 2017, 77, 3857–3869. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | Chr. | Cytoband | SNP Position | Transcripts Associated in cis | Gene Start | Gene End | p-Value |
|---|---|---|---|---|---|---|---|
| rs4873815 | 8 | 8q24.3 | 144796206 | ZNF707 | 144824516 | 144849514 | 8.92e−4 |
| 8q24.3 | NAPRT1 | 144728101 | 144731656 | 4.41e−4 | |||
| rs12976454 | 19 | 19p13.3 | 3495971 | APBA3 | 3701771 | 3712673 | 3.40e−4 |
| rs11136002 | 8 | 8p21.3 | 22273027 | C8orf58 | 22513067 | 22517605 | 5.53e−4 |
| 8p21.3 | KIAA1967 | 22518202 | 22533920 | 9.19e−4 | |||
| 8p21.3 | RHOBTB2 | 22913059 | 22933655 | 3.12e−4 | |||
| TNFRSF10B | 22933598 | 22982637 | 4.03e−4 | ||||
| rs13259097 | 8 | 8p21.3 | 22189689 | RHOBTB2 | 22913059 | 22933655 | 3.87e−4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Chung, J.-Y.; Hwang, J.R.; Lee, Y.-Y.; Kim, T.-J.; Lee, J.-W.; Kim, B.-G.; Bae, D.-S.; Choi, C.H.; Hewitt, S.M. Identification of Candidate Genes Associated with Susceptibility to Ovarian Clear Cell Adenocarcinoma Using cis-eQTL Analysis. J. Clin. Med. 2020, 9, 1137. https://doi.org/10.3390/jcm9041137
Kim J, Chung J-Y, Hwang JR, Lee Y-Y, Kim T-J, Lee J-W, Kim B-G, Bae D-S, Choi CH, Hewitt SM. Identification of Candidate Genes Associated with Susceptibility to Ovarian Clear Cell Adenocarcinoma Using cis-eQTL Analysis. Journal of Clinical Medicine. 2020; 9(4):1137. https://doi.org/10.3390/jcm9041137
Chicago/Turabian StyleKim, Jihye, Joon-Yong Chung, Jae Ryoung Hwang, Yoo-Young Lee, Tae-Joong Kim, Jeong-Won Lee, Byoung-Gie Kim, Duk-Soo Bae, Chel Hun Choi, and Stephen M. Hewitt. 2020. "Identification of Candidate Genes Associated with Susceptibility to Ovarian Clear Cell Adenocarcinoma Using cis-eQTL Analysis" Journal of Clinical Medicine 9, no. 4: 1137. https://doi.org/10.3390/jcm9041137
APA StyleKim, J., Chung, J. -Y., Hwang, J. R., Lee, Y. -Y., Kim, T. -J., Lee, J. -W., Kim, B. -G., Bae, D. -S., Choi, C. H., & Hewitt, S. M. (2020). Identification of Candidate Genes Associated with Susceptibility to Ovarian Clear Cell Adenocarcinoma Using cis-eQTL Analysis. Journal of Clinical Medicine, 9(4), 1137. https://doi.org/10.3390/jcm9041137