Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Cell Culture
2.3. Cell Viability Assays
2.4. Statistical Analysis and the Half Maximal Inhibitory Concentration (IC50) Calculations
3. Results
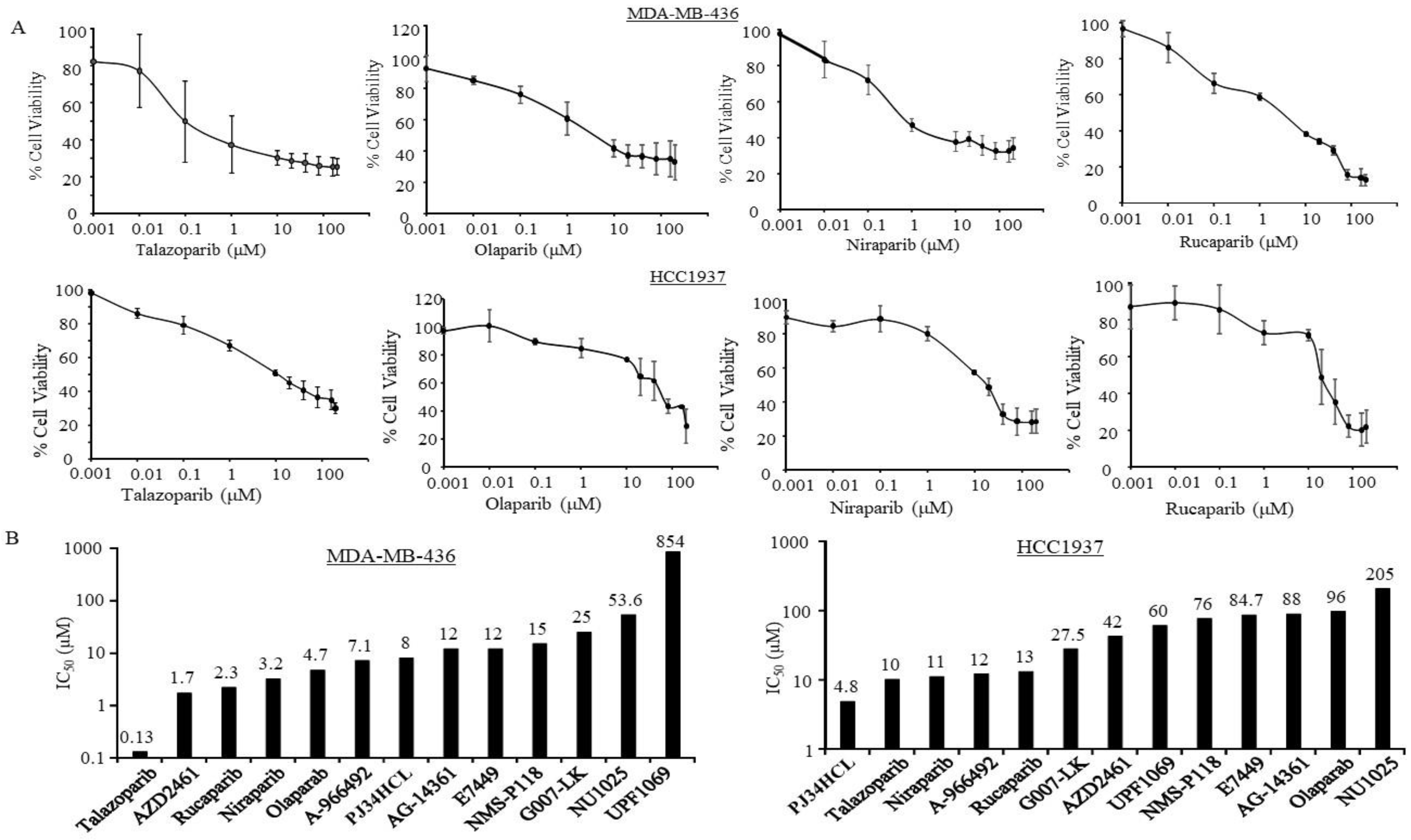
3.1. Sensitivity of PARPi in TNBC Cells with Germline BRCA Mutations
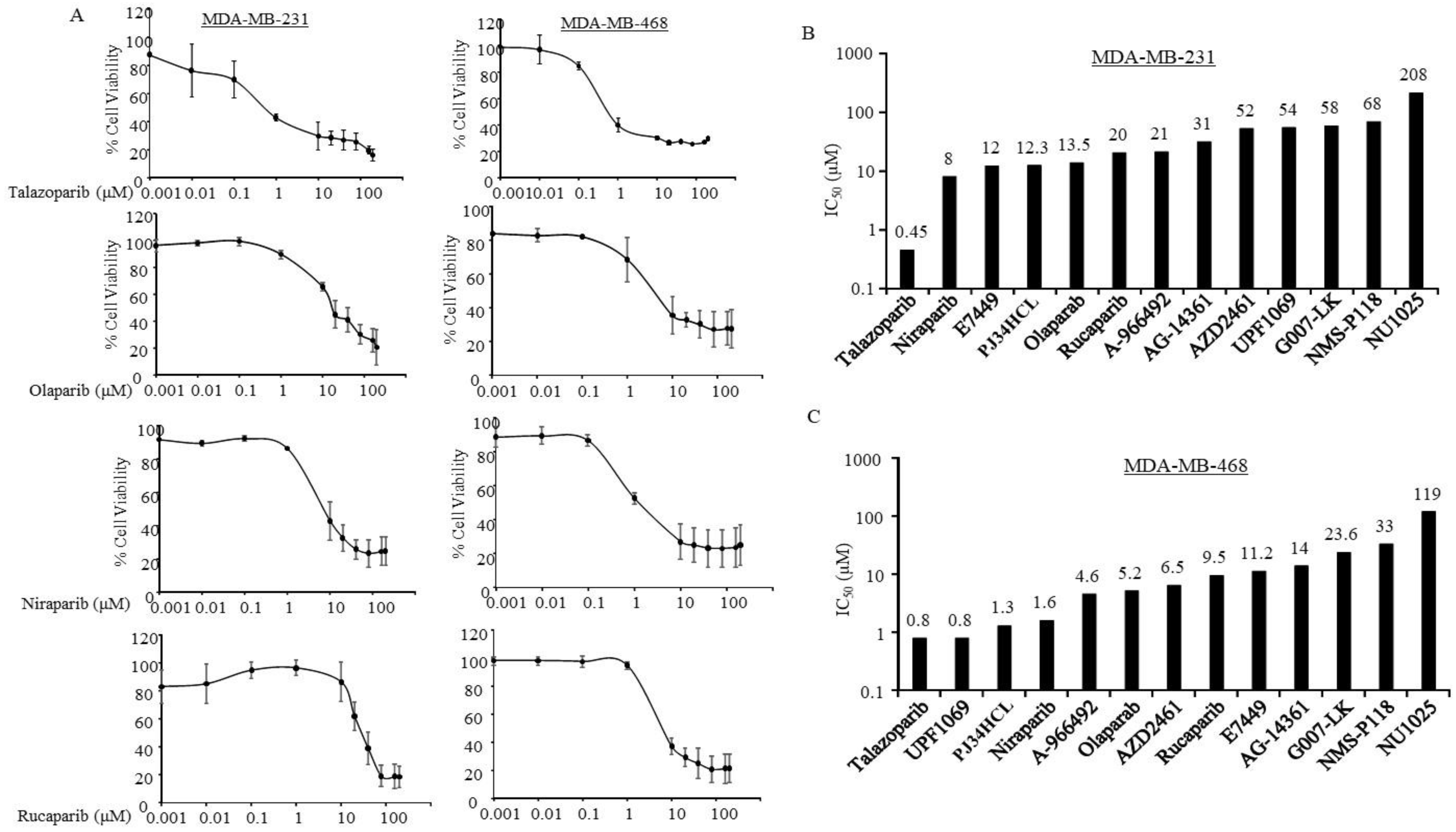
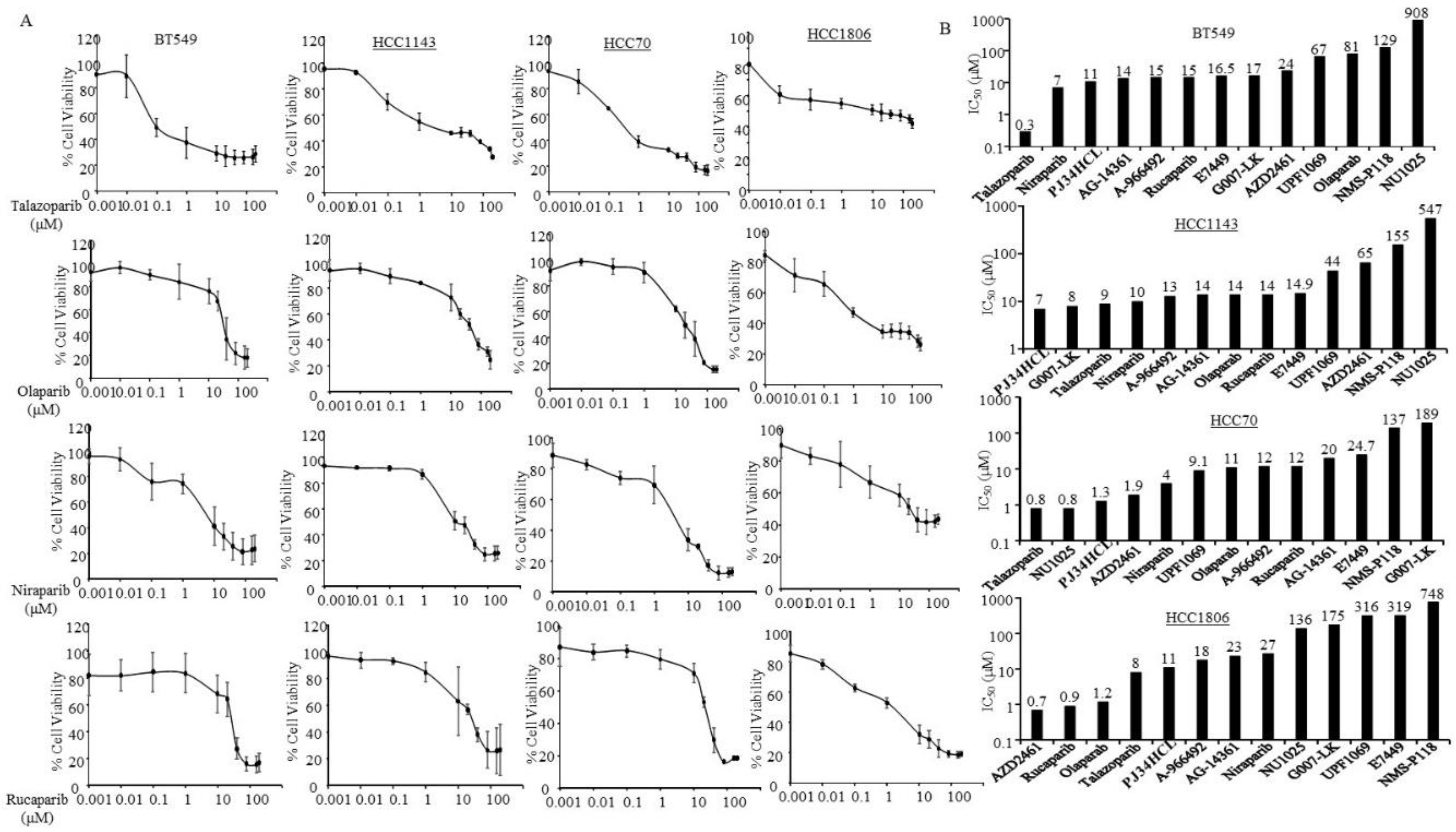
3.2. Sensitivity of PARPi in TNBC Cells without Germline BRCA Mutations
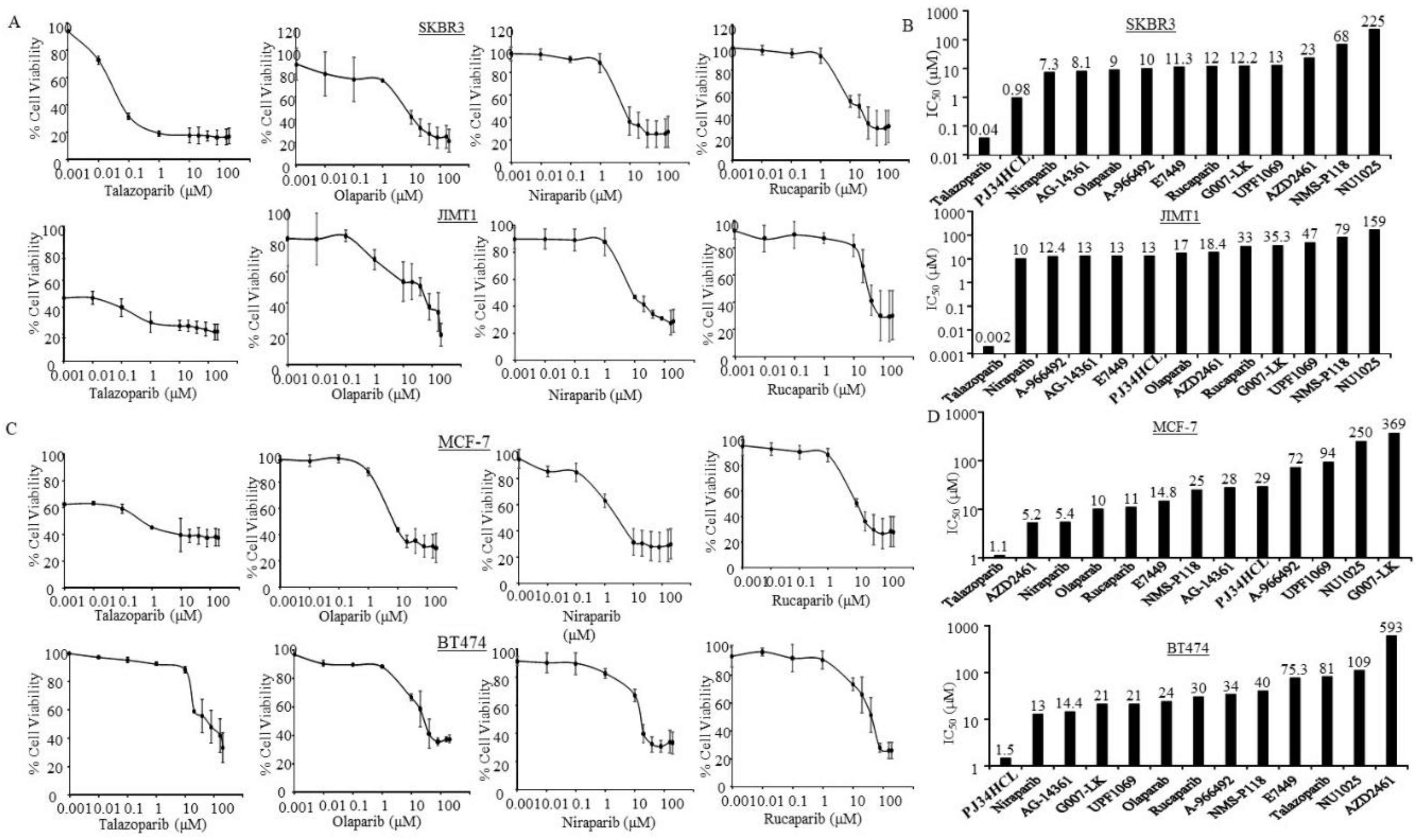
3.3. Sensitivity of PARPi in ER-Negative and HER2-Positive Breast Cancer Cells
3.4. Sensitivity of PARPi in ER-Positive Breast Cancer Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological Types of Breast Cancer: How Special Are They? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Fish, K.; Shema, S.J.; Clarke, C.A. Lifetime Risks of Specific Breast Cancer Subtypes among Women in Four Racial/ethnic Groups. Breast Cancer Res. 2010, 12, R99. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sarkissyan, M.; Elshimali, Y.; Vadgama, J.V. Triple Negative Breast Tumors in African-American and Hispanic/Latina Women Are High in CD44+, Low in CD24+, and Have Loss of PTEN. PLoS ONE 2013, 8, 78259. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Khan, H.; Chillar, R.; Vadgama, J.V. Prognostic Value of Plasma HER-2/neu in African American and Hispanic Women with Breast Cancer. Int. J. Oncol. 1999, 14, 1021–1058. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, P.R.; Mayer, I.A.; Mernaugh, R. Resistance to Trastuzumab in Breast Cancer. Clin. Cancer Res. 2009, 15, 7479–7491. [Google Scholar] [CrossRef]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory; Gumbs, C.; Micklem, G. Identification of the Breast Cancer Susceptibility Gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary Breast and Ovarian Cancer due to Mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Malone, K.E.; Daling, J.R.; Doody, D.R.; Hsu, L.; Bernstein, L.; Coates, R.J.; Marchbanks, P.A.; Simon, M.S.; McDonald, J.A.; Norman, S.A.; et al. Prevalence and Predictors of BRCA1 and BRCA2 Mutations in a Population-Based Study of Breast Cancer in White and Black American Women Ages 35 to 64 Years. Cancer Res. 2006, 66, 8297–8308. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.; Tonin, P.; Durocher, F.; Morgan, K.; Rommens, J.; Gingras, S.; Samson, C.; Leblanc, J.F.; Bélanger, C.; Dion, F.; et al. Common origins of BRCA1 Mutations in Canadian Breast and Ovarian Cancer Families. Nat. Genet. 1994, 8, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wu, Y.; Subbarao, M.; Bhat, H.; Chillar, R.; Vadgama, J.V. Mutation Analysis of BRCA1 Gene in African-American Patients with Breast Cancer. J. Natl. Med. Assoc. 2000, 92, 29–35. [Google Scholar] [PubMed]
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association Between BRCA Status and Triple-Negative Breast Cancer: A Meta-Analysis. Front. Pharmacol. 2018, 9, 909. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural Basis for DNA Damage-Dependent Poly (ADP-Ribosyl) Ation by Human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and Their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef]
- Inbar-Rozensal, D.; Castiel, A.; Visochek, L.; Castel, D.; Dantzer, F.; Izraeli, S.; Cohen-Armon, M. A Selective Eradication of Human Nonhereditary Breast Cancer Cells by Phenanthridine-Derived polyADP-Ribose Polymerase Inhibitors. Breast Cancer Res. 2009, 11, R78. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.E.; Jensen, R.A.; Obermiller, P.S.; Page, D.L.; Holt, J.T. Decreased Expression of BRCA1 Accelerates Growth and Is Often Present during Sporadic Breast Cancer Progression. Nat. Genet. 1995, 9, 444–450. [Google Scholar] [CrossRef]
- Taylor, J.; Lymboura, M.; Pace, P.; A’hern, R.P.; Desai, A.J.; Shousha, S.; Coombes, R.C.; Ali, S. An Important Role for BRCA1 in Breast Cancer Progression Is Indicated by Its Loss in a Large Proportion of Non-Familial Breast Cancers. Int. J. Cancer 1998, 79, 334–342. [Google Scholar] [CrossRef]
- Rhiem, K.; Todt, U.; Wappenschmidt, B.; Klein, A.; Wardelmann, E.; Schmutzler, R.K. Sporadic Breast Carcinomas with Somatic BRCA1 Gene Deletions Share Genotype/Phenotype Features with Familial Breast Carcinomas. Anticancer Res. 2010, 30, 3445–3449. [Google Scholar] [PubMed]
- Elstrodt, F.; Hollestelle, A.; Nagel, J.H.; Gorin, M.; Wasielewski, M.; van den Ouweland, A.; Merajver, S.D.; Ethier, S.P.; Schutte, M. BRCA1 Mutation Analysis of 41 Human Breast Cancer Cell Lines Reveals Three New Deleterious Mutants. Cancer Res. 2006, 66, 41–45. [Google Scholar] [CrossRef]
- PARP Inhibitor Review - Selleck Chemicals. Available online: https://www.selleckchem.com/PARP.html (accessed on 7 March 2020).
- Węsierska-Gądek, J.; Mauritz, M.; Mitulovic, G.; Cupo, M. Differential Potential of Pharmacological PARP Inhibitors for Inhibiting Cell Proliferation and Inducing Apoptosis in Human Breast Cancer Cells. J. Cell Biochem. 2015, 116, 2824–2839. [Google Scholar] [CrossRef]
- Antolín, A.A.; Jalencas, X.; Yélamos, J.; Mestres, J. Identification of Pim Kinases as Novel Targets for PJ34 with Confounding Effects in PARP Biology. ACS Chem. Biol. 2012, 7, 1962–1967. [Google Scholar] [CrossRef]
- Shirley, M. Rucaparib: A Review in Ovarian Cancer. Target Oncol. 2019, 14, 237–246. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Ke, Y.; Zhang, J.; Lv, X.; Zeng, X.; Ba, X. Novel Insights into PARPs in Gene Expression: Regulation of RNA Metabolism. Cell. Mol. Life Sci. 2019, 76, 3283–3299. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Giriat, I.; Schmitt, A.; De Lange, T. Tankyrase, a Poly (ADP-Ribose) Polymerase at Human Telomeres. Science 1998, 282, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Exman, P.; Barroso-Sousa, R.; Tolaney, S.M. Evidence to Date: Talazoparib in the Treatment of Breast Cancer. OncoTargets Ther. 2019, 12, 5177. [Google Scholar] [CrossRef] [PubMed]
- American Type Culture Collection. Available online: https://www.atcc.org/en/Products/Cells_and_Microorganisms/Cell_Lines.aspx (accessed on 7 March 2020).
- Tanner, M.; Kapanen, A.I.; Junttila, T.; Raheem, O.; Grenman, S.; Elo, J.; Elenius, K.; Isola, J. Characterization of a Novel Cell Line Established from a Patient with Herceptin-Resistant Breast Cancer. Mol. Cancer Ther. 2004, 3, 1585–1592. [Google Scholar]
- Matulonis, U.A. PARP Inhibitors in BRCA-Related Ovarian Cancer—And Beyond! Available online: http://www.ascopost.com/issues/november-25-2017/parp-inhibitors-in-brca-related-ovarian-cancer-and-beyond/ (accessed on 8 June 2018).
- AstraZeneca. AstraZeneca Press Release: LYNPARZATM Approved by the US Food and Drug Administration for the Treatment of Advanced Ovarian Cancer in Patients with Germline BRCA-Mutations. Available online: https://www.astrazeneca-us.com/media/press-releases/2014/lynparza-approved-by-the-us-fda-20141219.html# (accessed on 12 April 2018).
- The U.S. Food and Drug Administration. FDA News Release: FDA Approves Talazoparib for gBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm623540.htm (accessed on 11 January 2019).
- The U.S. Food and Drug Administration. FDA News Release: FDA Approves First Treatment for Breast Cancer with a Certain Inherited Genetic Mutation. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm592347.htm (accessed on 12 April 2018).
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef]
- Joshi, P.M.; Sutor, S.L.; Huntoon, C.J.; Karnitz, L.M. Ovarian Cancer-Associated Mutations Disable Catalytic Activity of CDK12, a Kinase That Promotes Homologous Recombination Repair and Resistance to Cisplatin and Poly (ADP-Ribose) Polymerase Inhibitors. J. Biol. Chem. 2014, 289, 9247–9253. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 Complex Maintains Genomic Stability via Regulation of Expression of DNA Damage Response Genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef]
- Naidoo, K.; Wai, P.T.; Maguire, S.L.; Daley, F.; Haider, S.; Kriplani, D.; Campbell, J.; Mirza, H.; Grigoriadis, A.; Tutt, A.; et al. Evaluation of CDK12 Protein Expression as a Potential Novel Biomarker for DNA Damage Response Targeted Therapies in Breast Cancer. Mol Cancer Ther. 2018, 17, 306–315. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The Clonal and Mutational Evolution Spectrum of Primary Triple-Negative Breast Cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; De Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on in Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect Is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat. Med. 2017, 23, 517. [Google Scholar] [CrossRef]
- McGrail, D.J.; Lin, C.C.-J.; Garnett, J.; Liu, Q.; Mo, W.; Dai, H.; Lu, Y.; Yu, Q.; Ju, Z.; Yin, J.; et al. Improved Prediction of PARP Inhibitor Response and Identification of Synergizing Agents through Use of a Novel Gene Expression Signature Generation Algorithm. NPJ Syst. Biol. Appl. 2017, 3, 1–12. [Google Scholar] [CrossRef]
- Kraus, W.L. Transcriptional Control by PARP-1: Chromatin Modulation, Enhancer-Binding, Coregulation, and Insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARP Inhibitors | PARP1 | PARP2 | PARP3 | PARP5a/TNKS1 ^ | PARP5b/TNKS2 § |
|---|---|---|---|---|---|
| A-966492 [27] | ++++ | +++ | |||
| AG-14361 [27] | ++ | ||||
| AZD2461 [27] | ++ | +++ | + | + | |
| E7449 [27] | ++++ | ++++ | |||
| G007-LK [27] | + | + | |||
| Niraparib [27] | +++ | +++ | |||
| NMS-P118 [27] | ++ | ||||
| NU1025 [28] | + | ||||
| Olaparib [27] | ++ | ++++ | |||
| PJ34-HCl [29] | ++ | ++ | |||
| Rucaparib [30] | +++ | +++ | +++ | ||
| Talazoparib [27] | ++++ | ||||
| UPF-1069 [27] | + | ++ |
| Cell Line | Subtype | BRCA Mutations | Derivation | Disease |
|---|---|---|---|---|
| MDA-MB-231 | TNBC-Mesenchymal-like | No | 51 years Caucasian female | Adenocarcinoma |
| MDA-MB-436 | TNBC- Mesenchymal-like | Yes; 5396 + 1 g > A/truncated protein | 43 years Caucasian female | Adenocarcinoma |
| MDA-MB-468 | TNBC basal-like | No | 51 years Black female | Adenocarcinoma |
| MCF-7 | Luminal A | No | 69 years Caucasian female | Adenocarcinoma |
| HCC1143 | TNBC basal-like-1 | No | 52 years Caucasian female | TNM stage IIA, grade 3, primary ductal carcinoma |
| HCC1937 | TNBC basal-like-1 | Yes; 5382 insC/frameshift | 23 years Caucasian female | TNM stage IIB, grade 3, primary ductal carcinoma |
| BT474 | Luminal B | No | 60 years Caucasian female | Ductal carcinoma |
| BT549 | TNBC-Mesenchymal | No | 72 years Caucasian female | Ductal carcinoma |
| SKBR3 | HER2+ (sensitive to Herceptin) | No | 43 years Caucasian female | Ductal carcinoma |
| JIMT1 | HER2+ (resistant to trastuzumab) | No | 62 years Caucasian female | Adenocarcinoma |
| HCC70 | TNBC basal-like | No | 49 years Black female | TNM stage IIIA, grade 3, primary ductal carcinoma |
| HCC1806 | TNBC basal-like | No | 60 years Black female | TNM stage IIB, grade 2, primary acantholytic squamous cell carcinoma |
| Estimated IC50 (µM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Lines | MDA-MB436 | HCC1937 | MDA-MB231 | MDA-MB468 | BT549 | HCC1143 | HCC70 | HCC1806 | SKBR3 | JIMT1 | MCF7 | BT474 |
| PARPi | ||||||||||||
| A-966492 | 7.1 | 12 | 21 | 4.6 | 15 | 13 | 12 | 18 | 10 | 12.4 | 72 | 34 |
| AG-14361 | 12 | 88 | 31 | 14 | 14 | 14 | 20 | 23 | 8.1 | 13 | 28 | 14.4 |
| AZD2461 | 1.7 | 42 | 52 | 6.5 | 24 | 65 | 1.9 | 0.7 | 23 | 18.4 | 5.2 | 593 |
| E7449 | 12 | 84.7 | 12 | 11.2 | 16.5 | 14.9 | 24.7 | 319 | 11.3 | 13 | 14.8 | 75.3 |
| G007-LK | 25 | 27.5 | 58 | 23.6 | 17 | 8 | 189 | 175 | 12.2 | 35.3 | 369 | 21 |
| Niraparib | 3.2 | 11 | 8 | 1.6 | 7 | 10 | 4 | 27 | 7.3 | 10 | 5.4 | 13 |
| NMS-P118 | 15 | 76 | 68 | 33 | 129 | 155 | 137 | 748 | 68 | 79 | 25 | 40 |
| NU1025 | 53.6 | 205 | 208 | 119 | 908 | 547 | 0.8 | 136 | 225 | 159 | 250 | 109 |
| Olaparib | 4.7 | 96 | 13.5 | 5.2 | 81 | 14 | 11 | 1.2 | 9 | 17 | 10 | 24 |
| PJ34HCL | 8 | 4.8 | 12.3 | 1.3 | 11 | 7 | 1.3 | 11 | 0.98 | 13 | 29 | 1.5 |
| Rucaparib | 2.3 | 13 | 20 | 9.5 | 15 | 14 | 12 | 0.9 | 12 | 33 | 11 | 30 |
| Talazoparib | 0.13 | 10 | 0.45 | 0.8 | 0.3 | 9 | 0.8 | 8 | 0.04 | 0.002 | 1.1 | 81 |
| UPF1069 | 854 | 60 | 54 | 0.8 | 67 | 44 | 9.1 | 316 | 13 | 47 | 94 | 21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. https://doi.org/10.3390/jcm9040940
Keung MY, Wu Y, Badar F, Vadgama JV. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. Journal of Clinical Medicine. 2020; 9(4):940. https://doi.org/10.3390/jcm9040940
Chicago/Turabian StyleKeung, Man Yee, Yanyuan Wu, Francesca Badar, and Jaydutt V. Vadgama. 2020. "Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status" Journal of Clinical Medicine 9, no. 4: 940. https://doi.org/10.3390/jcm9040940
APA StyleKeung, M. Y., Wu, Y., Badar, F., & Vadgama, J. V. (2020). Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. Journal of Clinical Medicine, 9(4), 940. https://doi.org/10.3390/jcm9040940