Serotonin-Secreting Neuroendocrine Tumours of the Pancreas




Abstract
:1. Introduction
2. Methods
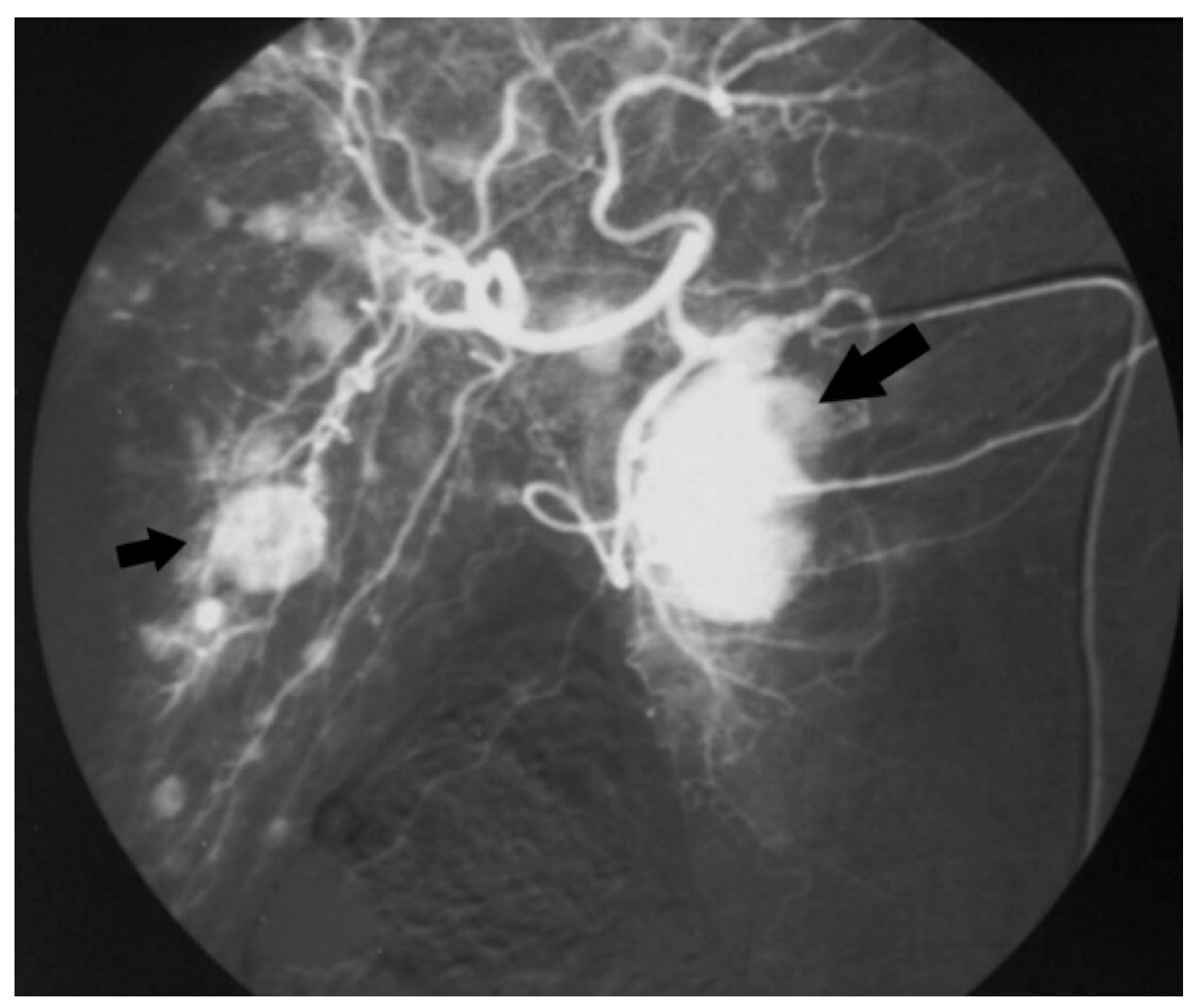
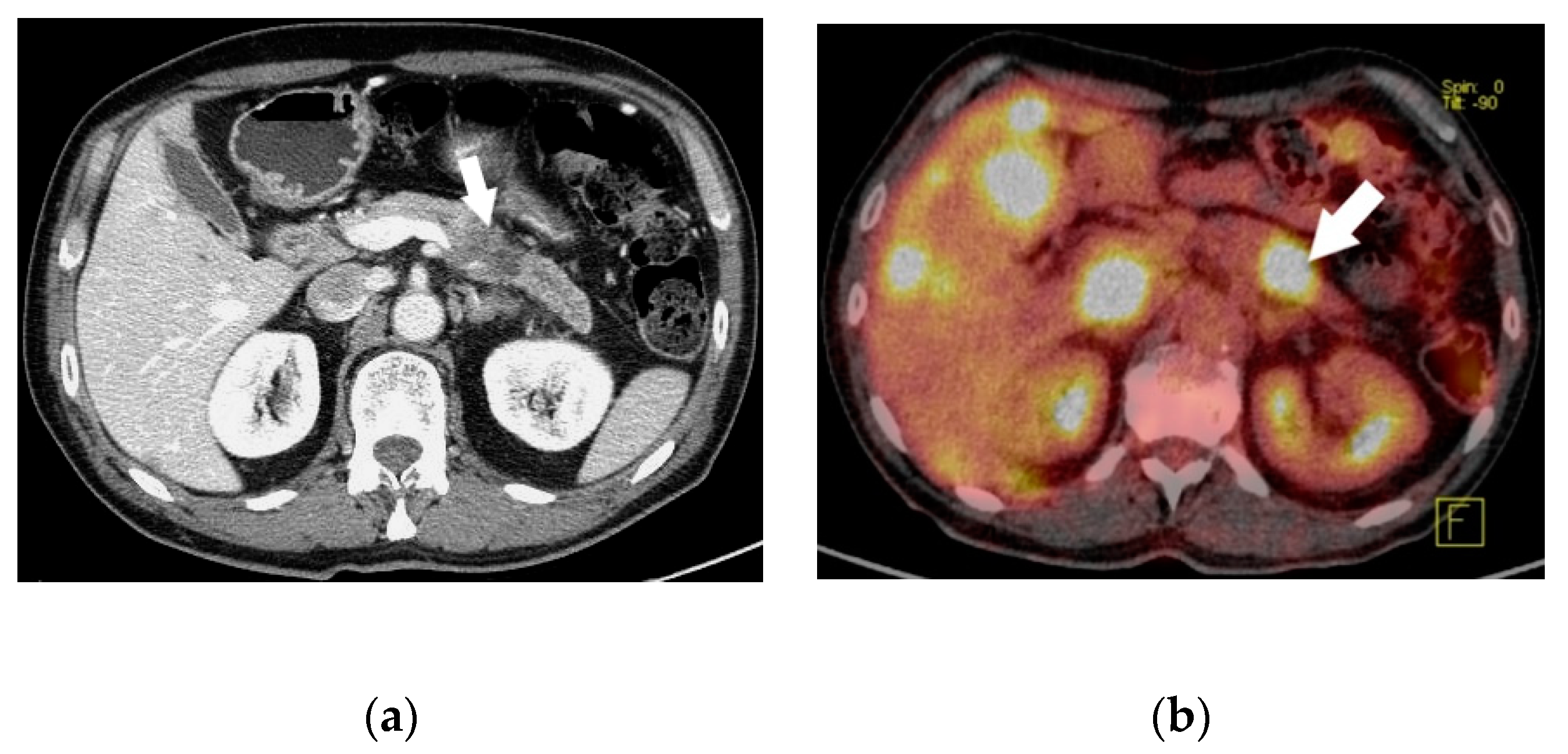
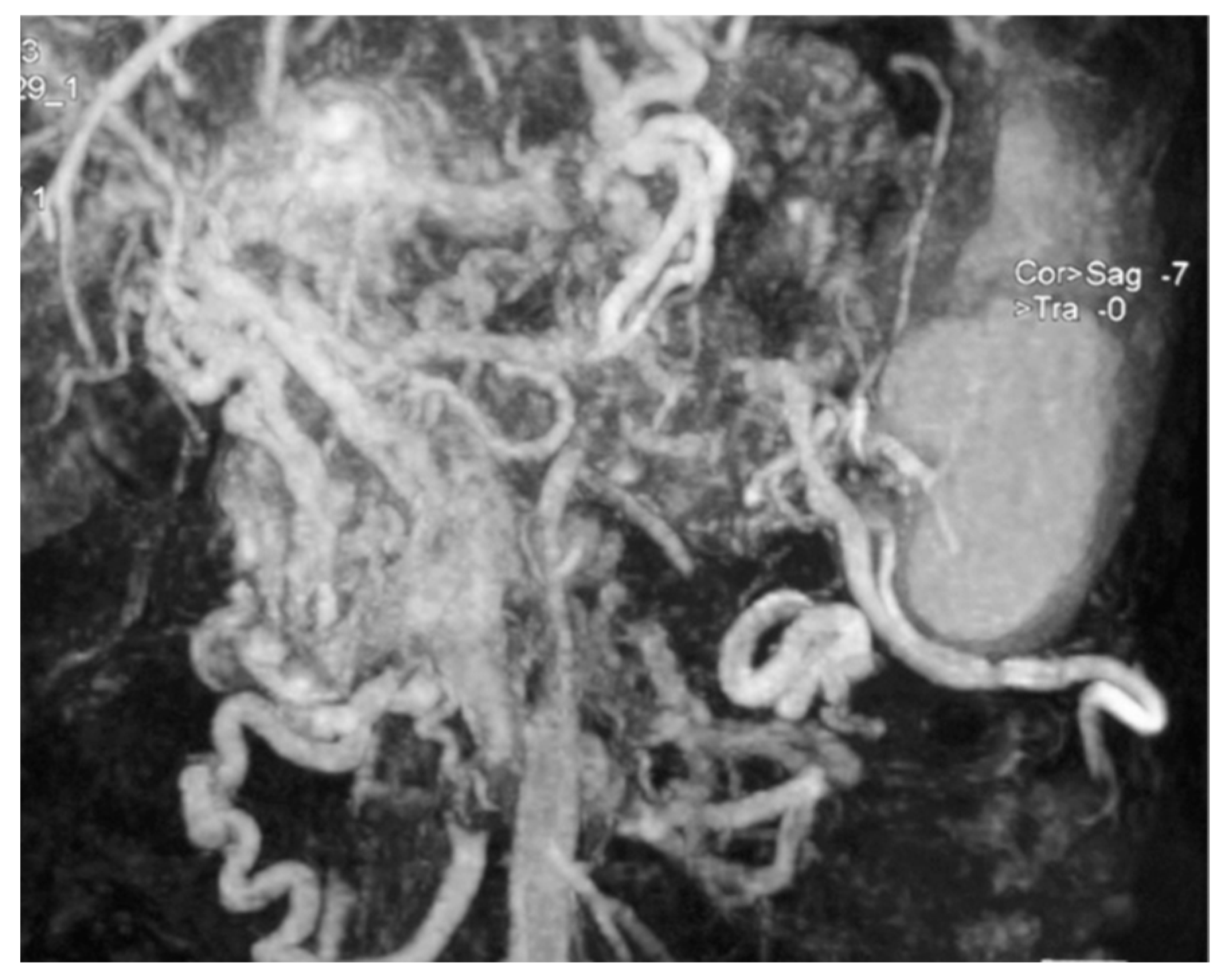
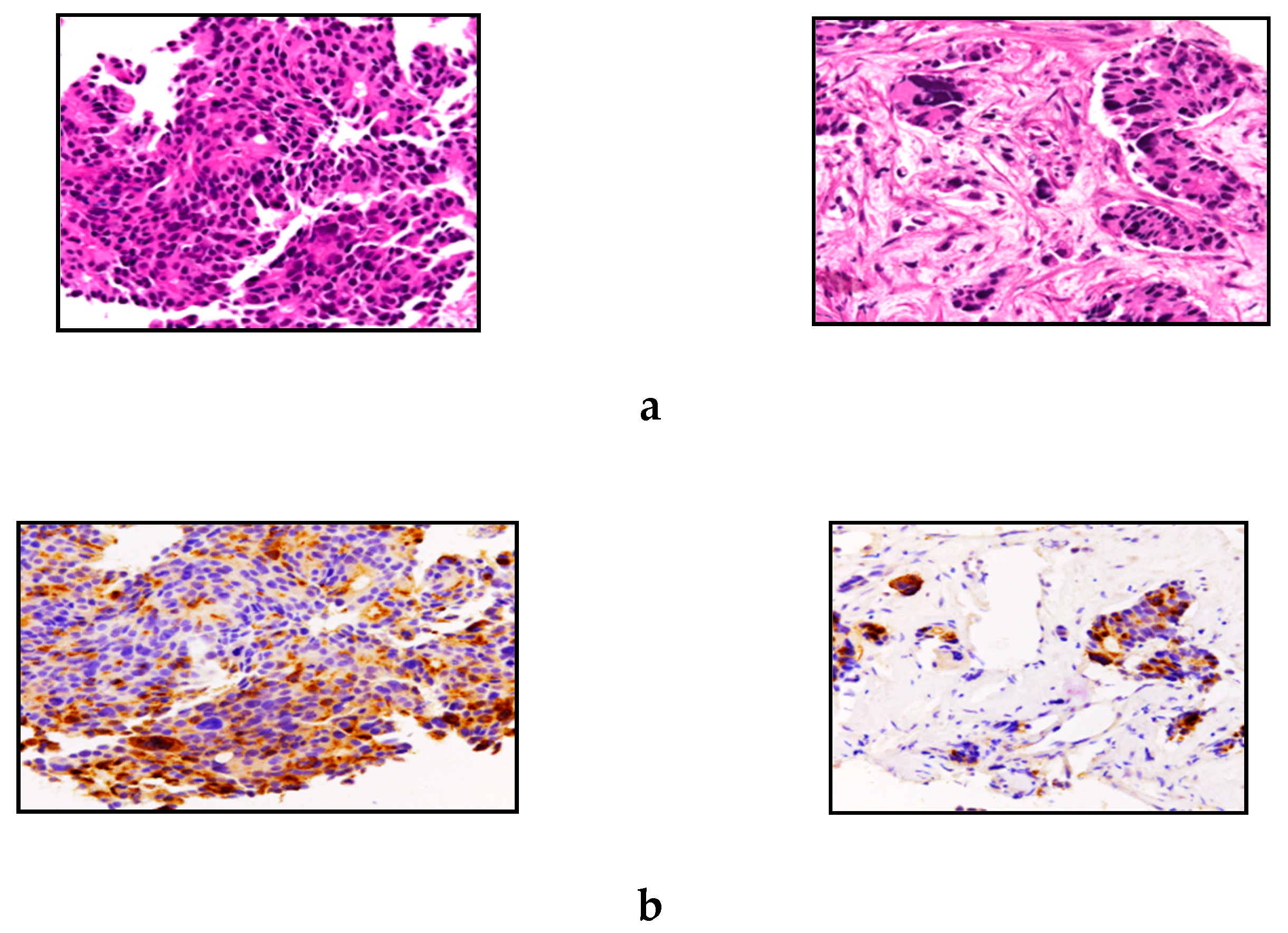
3. Results
3.1. Patient Characteristics and Laboratory Diagnosis
3.2. Histology and Immunohistochemical Features
3.3. Prognosis and Follow-Up
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Williams, E.D.; Sanders, M. The classification of carcinoid tumors. Lancet 1963, 1, 238–239. [Google Scholar] [CrossRef]
- Gustafsson, B.I. Small Intestinal Neuroendocrine Tumours. In A Century of Advances in Neuroendocrine Tumour Biology and Treatment; Modlin, I.M., Ed.; Felsenstein C.C.C.P.: Hannover, Germany, 2008; p. 103. [Google Scholar]
- Öberg, K. Non-Functioning Pancreatic Endocrine Tumour. In A Century of Advances in Neuroendocrine Tumour Biology and Treatment; Modlin, I.M., Ed.; Felsenstein C.C.C.P.: Hannover, Germany, 2008; p. 87. [Google Scholar]
- Wilson, R.W.; Gal, A.A.; Cohen, C.; DeRose, P.B.; Millikan, W.J. Serotonin immunoreactivity in pancreatic endocrine neoplasms (carcinoid tumors). Mod. Pathol. 1991, 4, 727–732. [Google Scholar] [PubMed]
- Bilimoria, K.Y.; Tomlinson, J.S.; Merkow, R.P.; Stewart, A.K.; Ko, C.Y.; Talamonti, M.S.; Bentrem, D.J. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: Analysis of 9,821 patients. J. Gastrointest. Surg. 2007, 11, 1460–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, S.; Franzi, F.; Albarello, L.; Schmitt, A.; Bernasconi, B.; Tibiletti, M.G.; Finzi, G.; Placidi, C.; Perren, A.; Capella, C. Serotonin-producing enterochromaffin cell tumors of the pancreas: Clinicopathologic study of 15 cases and comparison with intestinal enterochromaffin cell tumors. Pancreas 2011, 40, 883–895. [Google Scholar] [CrossRef]
- Massironi, S.; Partelli, S.; Petrone, M.C.; Zilli, A.; Conte, D.; Falconi, M.; Arcidiacono, P.G. Endoscopic ultrasound appearance of pancreatic serotonin-staining neuroendocrine neoplasms. Pancreatology 2018, 18, 792–798. [Google Scholar] [CrossRef]
- Maurer, C.A.; Baer, H.U.; Dyong, T.H.; Mueller-Garamvoelgyi, E.; Friess, H.; Ruchti, C.; Reubi, J.C.; Büchler, M.W. Carcinoid of the pancreas: Clinical characteristics and morphological features. Eur. J. Cancer 1996, 32, 1109–1116. [Google Scholar] [CrossRef]
- Dollinger, M.R.; Ratner, L.H.; Shamoian, C.A.; Blackboume, B.D. Carcinoid syndrome associated with pancreatic tumors. Arch. Int. Med. 1967, 120, 575–580. [Google Scholar] [CrossRef]
- Eusebi, V.; Capella, C.; Bondi, A.; Sessa, F.; Vezzadini, P.; Mancini, A.M. Endocrine-paracrine cells in pancreatic exocrine carcinomas. Histopathology 1981, 5, 599–613. [Google Scholar] [CrossRef]
- Digestive System Tumours. WHO Classification of Tumours, 5th ed.; WHO Classification of Tumours Editorial Board, Ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2019. [Google Scholar]
- Rindi, G.; Klöppel, G.; Alhman, X.; Caplin, M.; Couvelard, A.; De Herder, W.W.; Erikssson, B.; Falchetti, A.; Falconi, M.; Komminoth, P.; et al. TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch 2006, 449, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97, 934–959. [Google Scholar] [CrossRef]
- Soga, J. Carcinoids of the pancreas: An analysis of 156 cases. Cancer 2005, 104, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Zandee, W.T.; van Adrichem, R.C.; Kamp, K.; Feelders, R.A.; van Velthuysen, M.F.; de Herder, W.W. Incidence and prognostic value of serotonin secretion in pancreatic neuroendocrine tumours. Clin. Endocrinol (Oxf.) 2017, 87, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Osamura, R.Y. Serotonin-secreting tumours. In Pathology and Genetics of Tumours of Endocrine Organs. World Health Organization Classification of Tumours, 3rd ed.; DeLellis, R.A., Ed.; IARC Press: Lyon, France, 2004; p. 198. [Google Scholar]
- Yeo, C.J.; Couse, N.F.; Zinner, M.J. Serotonin and substance P stimulate intestinal secretion in the isolated perfused ileum. Surgery 1989, 105, 86–92. [Google Scholar]
- von der Ohe, M.R.; Camilleri, M.; Kvols, L.K.; Thomforde, G.M. Motor dysfunction of the small bowel and colon in patients with the carcinoid syndrome and diarrhea. N. Engl. J. Med. 1993, 329, 1073–1078. [Google Scholar] [CrossRef]
- Vinik, A.I.; Gonin, J.; England, B.G.; Jackson, T.; McLeod, M.K.; Cho, K. Plasma substance-P in neuroendocrine tumors and idiopathic flushing: The value of pentagastrin stimulation tests and the effects of somatostatin analog. J. Clin. Endocrinol. Metab. 1990, 70, 1702–1709. [Google Scholar] [CrossRef]
- Kapran, Y.; Bauersfeld, J.; Anlauf, M.; Sipos, B.; Klöppel, G. Multihormonality and entrapment of islets in pancreatic endocrine tumors. Virchows Arch 2006, 448, 394–398. [Google Scholar] [CrossRef]
- Rindi, G. Classification of neuroendocrine tumours. In Handbook of Gastroenteropancreatic and Thoracic Neuroendocrine Tumours, 1st ed.; Caplin, M., Ed.; BioScientifica: Bristol, UK, 2011; p. 35. [Google Scholar]
- Ohta, Y.; Kosaka, Y.; Kishimoto, N.; Wang, J.; Smith, S.B.; Honig, G.; Kim, H.; Gasa, R.M.; Neubauer, N.; Liou, A.; et al. Convergence of the insulin and serotonin programs in the pancreatic β-cell. Diabetes 2011, 60, 3208–3216. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; el Attar, A.; Domenico, D.R.; Kim, K.; Howard, J.M. Carcinoid tumors of the pancreas. Status report based on two cases and review of the world’s literature. Int. J. Pancreatol. 1998, 23, 153–164. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival oucomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Zandee, W.T.; Kamp, K.; van Adrichem, R.C.; Feelders, R.A.; de Herder, W.W. Limited value for urinary 5-HIAA excretion as prognostic marker in gastrointestinal neuroendocrine tumours. Eur. J. Endocrinol. 2016, 175, 361–366. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Obs | Gender/Age | Clinical Presentation | Carcinoid Syndrome | 24-h Urinary 5-HIAA * | Serum 5-HT * | Other Serum NE Markersand GI Hormones | |
|---|---|---|---|---|---|---|---|---|
| normal | elevated | |||||||
| 1 | 1986 | F/67 | Abdominal pain | Yes (flushing, diarrhoea) | n.a. | 5.1x | Gastrin, Glucagon, Calcitonin | NSE |
| 2 | 1995 | M/64 | Asymptomatic | No | 12.3x | 3.8x | Gastrin | NSE, Calcitonin |
| 3 | 1999 | M/69 | n.a. | n.a. | 5.2x | n.a. | NSE, Insulin, Gastrin, Calcitonin | no |
| 4 | 2002 | M/44 | Weight loss, dyspepsia | No diarrhoea | 1.8x | n.a. | NSE, Gastrin | CgA, Glucagon, Calcitonin |
| 5 | 2004 | F/44 | Cervical lymphadenopathy | No | 6.7x | n.a. | NSE, Insulin | CgA |
| 6 | 2010 | F/38 | Weight loss, jaundice, portal vein thrombosis, ascites, fatigue | Yes (flushing, diarrhoea) | 17.4x | 1.3x | NSE, SS, VIP, Calcitonin | CgA, Gastrin |
| 7 | 2011 | M/68 | Abdominal pain, weight loss, fatigue | No diarrhoea | 4.5x | 2.1x | Gastrin | CgA, NSE, Calcitonin |
| No. | Pancreatic Site Size (cm) | Distant Metastases | Biopsy | TNM Stage [12] | NET/NEC Ki67 | Immunohistochemistry | Other Therapies | Follow-Up (Months) | Status | |
|---|---|---|---|---|---|---|---|---|---|---|
| Positive | Negative | |||||||||
| 1 | Head 4.0 | Bilobar liver | Liver | T2 Nx M1 IV | NET n.a. | 5-HT 20–20–30% Grimelius | Insulin, Gastrin, PP | SS-A, CT a | 158 | DOD |
| 2 | Tail 2.5 | Bilobar liver | Liver | T2 Nx M1 IV | NET n.a. | CgA | n.a. | TACE | 12 | DOD |
| 3 | Body 4.0 | Bilobar liver, mediastinal LN | Liver | T2 N1 M1 IV | NET n.a. | CgA, Grimelius | n.a. | CT b | 29 | DOD |
| 4 | Tail 6.0 | Bilobar liver | Abdominal LN | T3 N1 M1 IV | NET 3.4% | CgA, Syn, NSE | 5-HT, Insulin, Gastrin, Glucagon, SS, PP, Calcitonin | SS-A, CT b, TAE, PRRT | 96 | DOD |
| 5 | Body 10.0 | Bilobar liver, cervical LN | Cervical LN | T4 N1 M1 IV | NEC n.a. | n.a. | n.a. | SS-A, CT c | 16 | DOD |
| 6 | Body 4.0 | Bilobar liver | Liver | T2 N1 M1 IV | NET 16% | CgA, Syn | n.a. | Biliary stent SS-A, CT d Liver MW Everolimus | 117 | AWD |
| 7 | Body 3.6 | Bilobar liver | Liver | T2 Nx M1 IV | NET 70% | 5-HT, CgA, Syn, Calcitonin | NSE, Insulin, Gastrin, Glucagon, SS, PP, VIP | CT e | 5 | DOD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanetto, A.C.; Fassan, M.; David, A.; Pasquali, C. Serotonin-Secreting Neuroendocrine Tumours of the Pancreas. J. Clin. Med. 2020, 9, 1363. https://doi.org/10.3390/jcm9051363
Milanetto AC, Fassan M, David A, Pasquali C. Serotonin-Secreting Neuroendocrine Tumours of the Pancreas. Journal of Clinical Medicine. 2020; 9(5):1363. https://doi.org/10.3390/jcm9051363
Chicago/Turabian StyleMilanetto, Anna Caterina, Matteo Fassan, Alina David, and Claudio Pasquali. 2020. "Serotonin-Secreting Neuroendocrine Tumours of the Pancreas" Journal of Clinical Medicine 9, no. 5: 1363. https://doi.org/10.3390/jcm9051363
APA StyleMilanetto, A. C., Fassan, M., David, A., & Pasquali, C. (2020). Serotonin-Secreting Neuroendocrine Tumours of the Pancreas. Journal of Clinical Medicine, 9(5), 1363. https://doi.org/10.3390/jcm9051363