The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome

 ,
,  ,
,  , and
, and 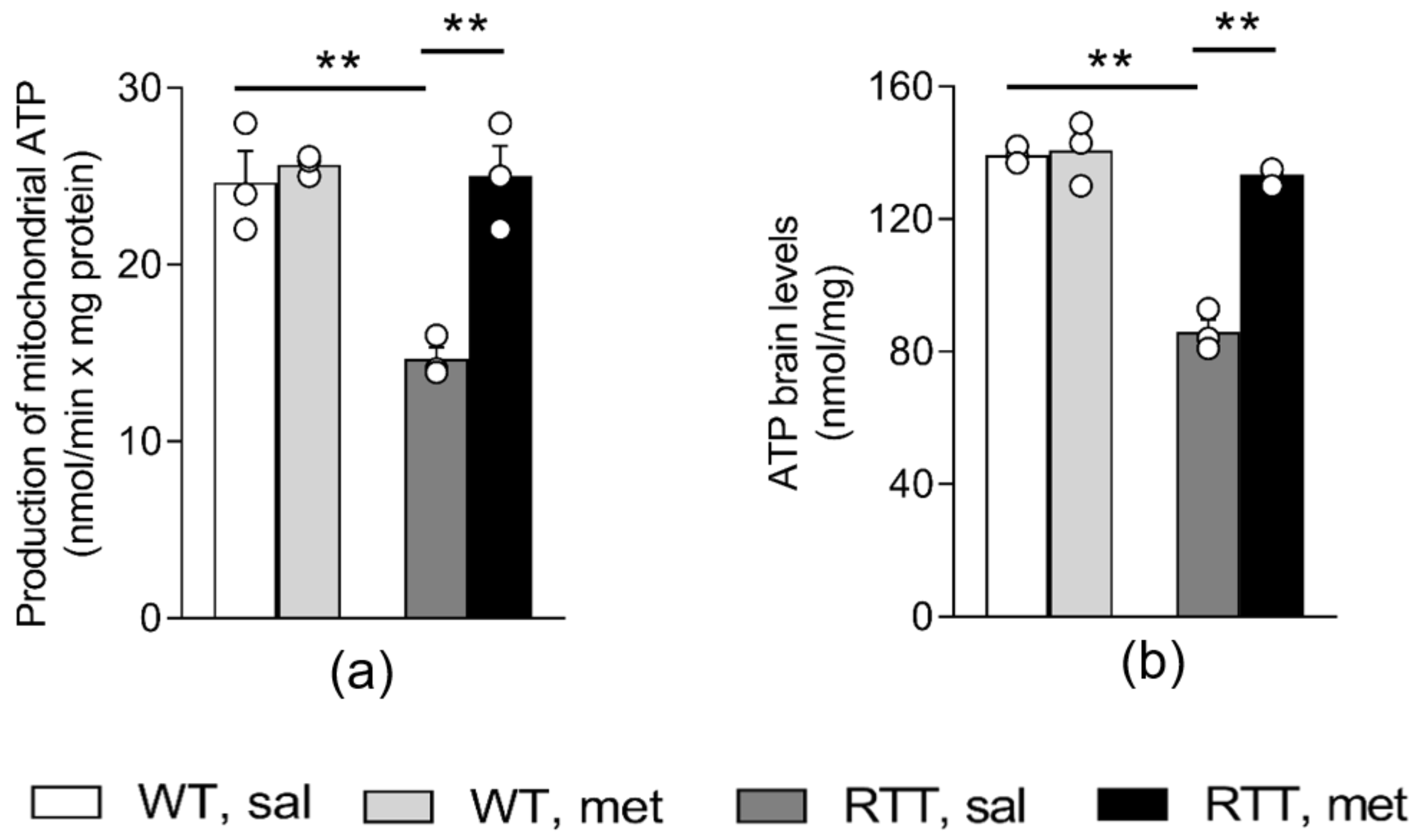{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Subjects
2.2. Genotyping
2.3. Drug Treatment and Experimental Design
2.4. Behavioral Assessments
2.4.1. Open Field Test
2.4.2. Dowel Test
2.4.3. General Health Evaluation
2.5. Mitochondrial Analyses
2.5.1. Brain Tissue Dissection and Mitochondria Isolation
2.5.2. Mitochondrial ATP Production via OXPHOS
2.5.3. Mouse Brain ATP Levels
2.5.4. MRC Complex Activities
2.6. Oxidative Stress Status Analyses
2.6.1. ROS Levels in Whole Blood by Electron Paramagnetic Resonance (EPR)
2.6.2. Total 4-hydroxy-2-trans-nonenal Protein Bound (HNE-Adducts) in RTT Mouse Brain
2.7. Western Blot Analyses
2.8. Statistical Analyses
3. Results
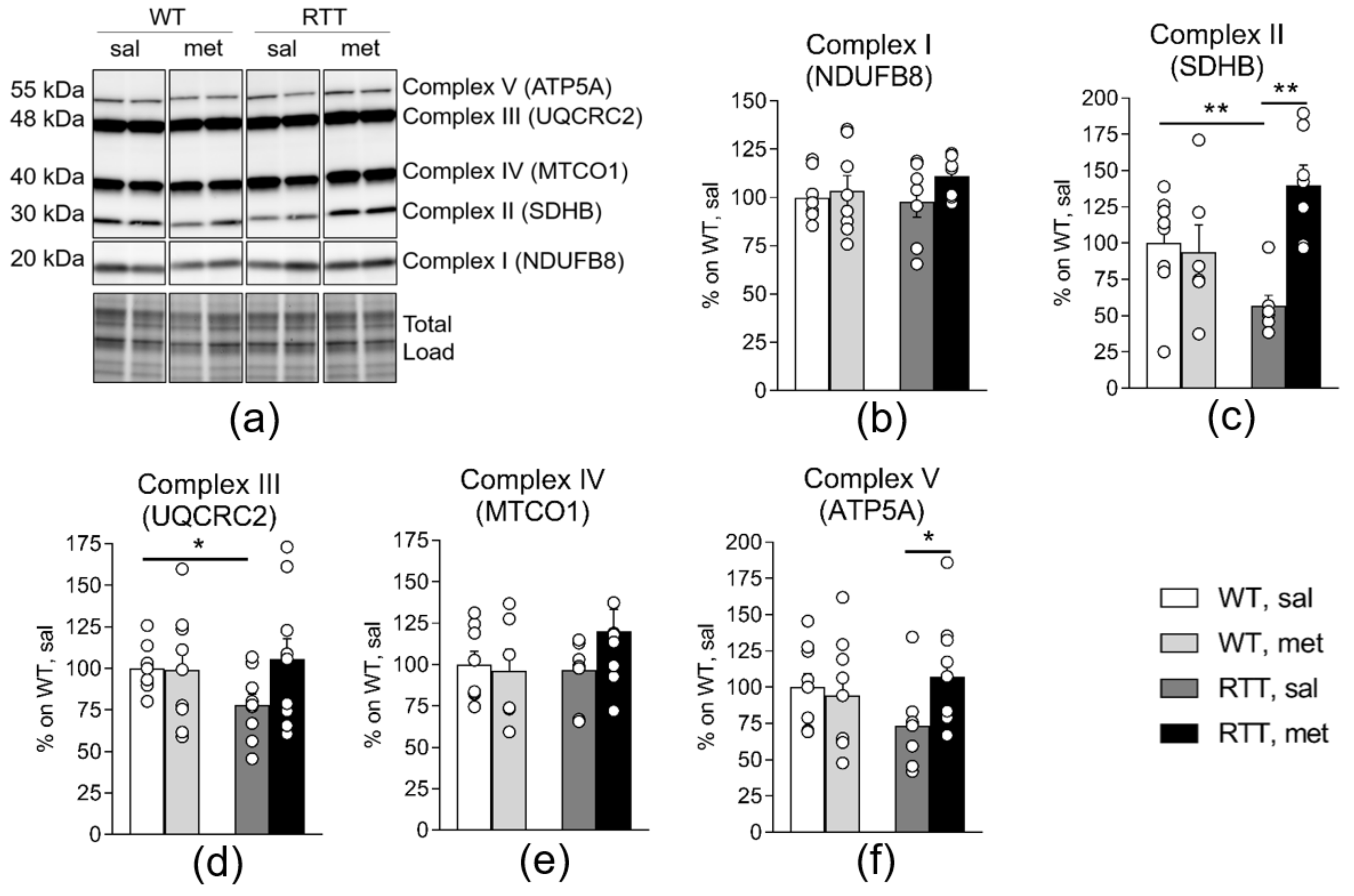
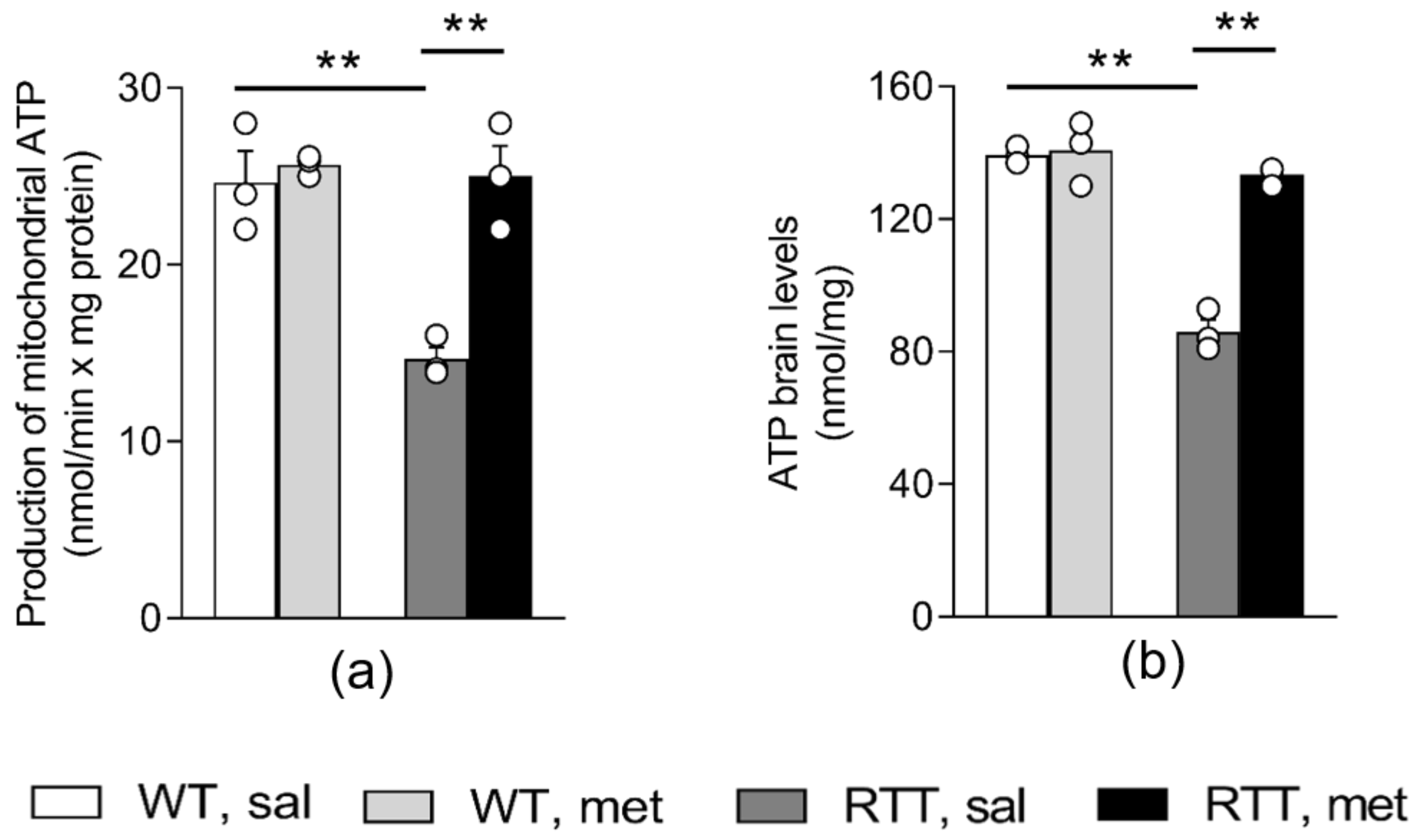
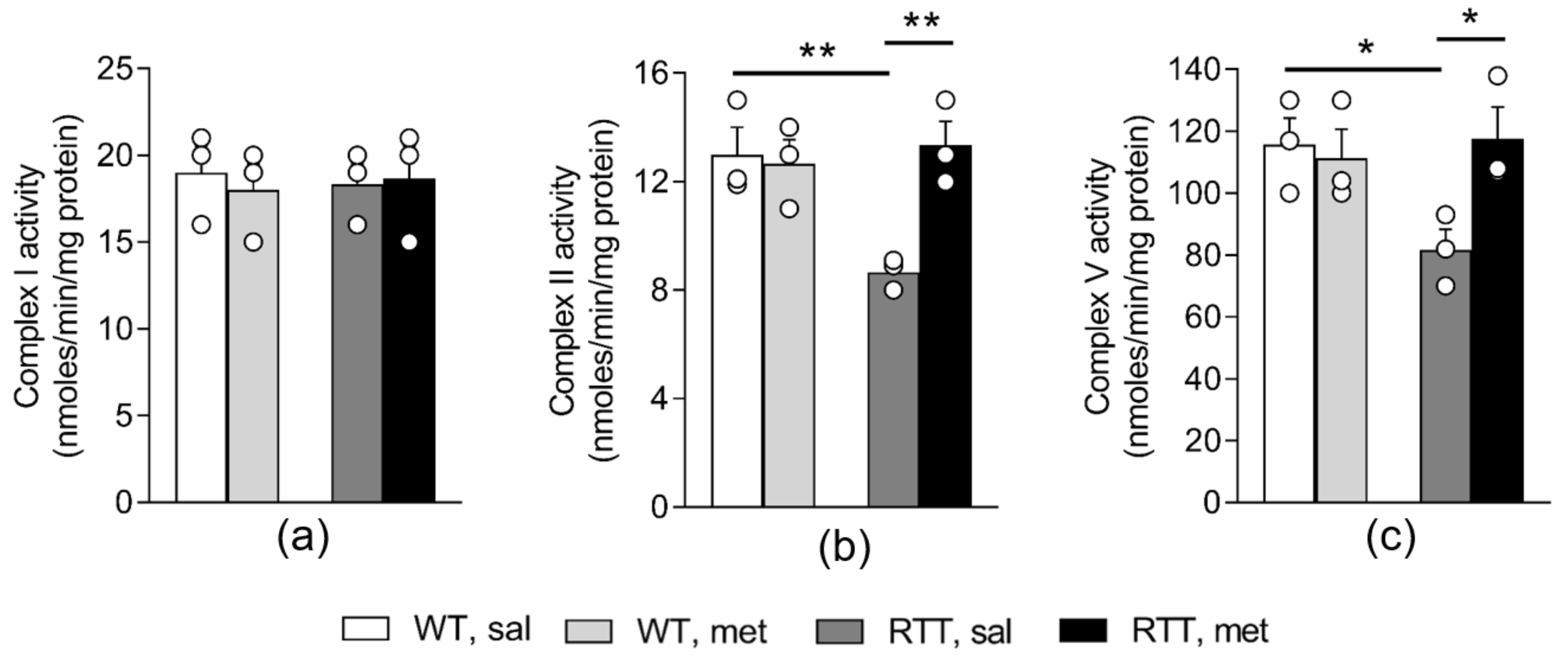
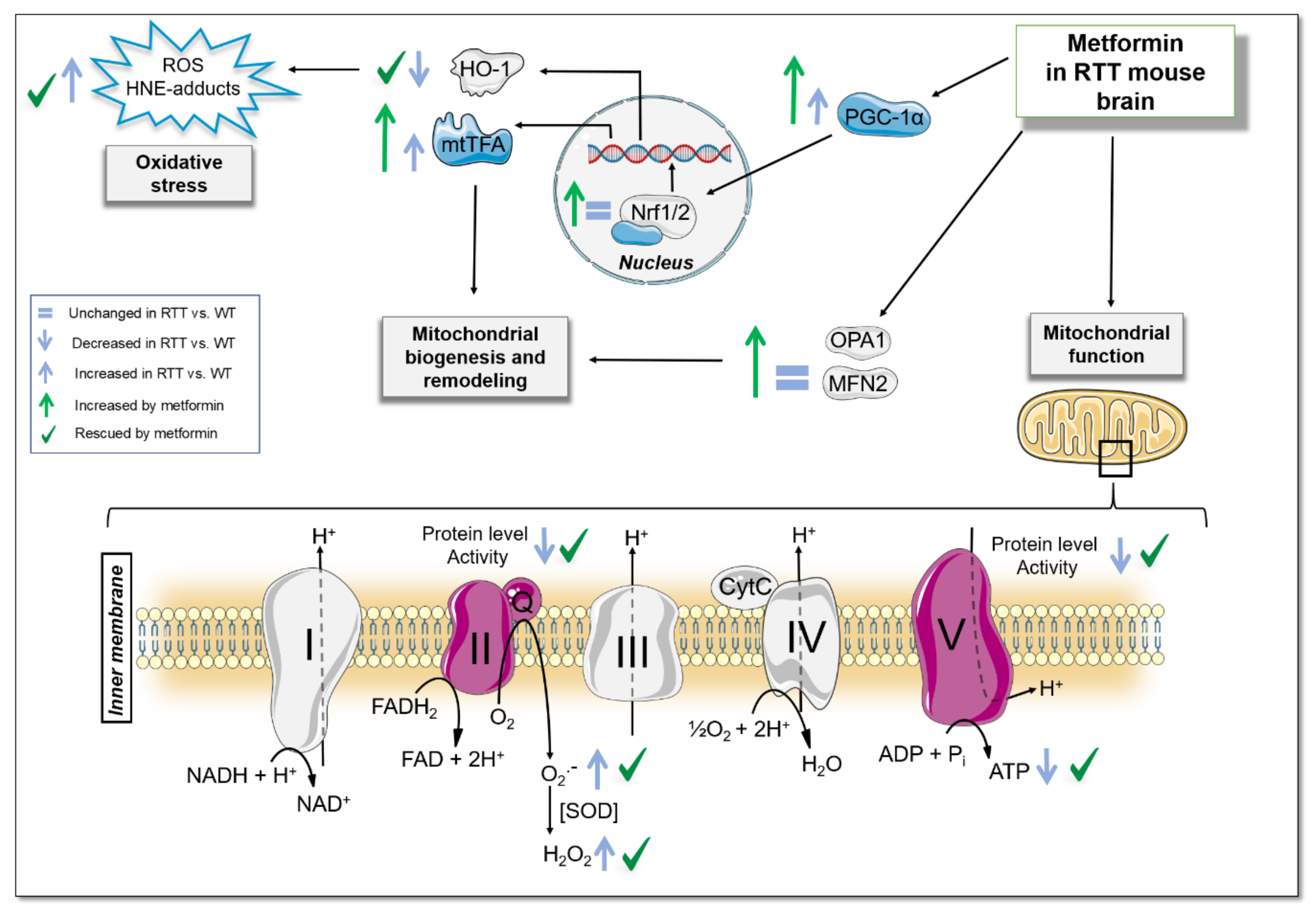
3.1. A 10-Day Long Metformin Treatment at 100 mg/kg Dose Rescues the Aberrant Mitochondrial Bioenergetics in the Brain of RTT Mice
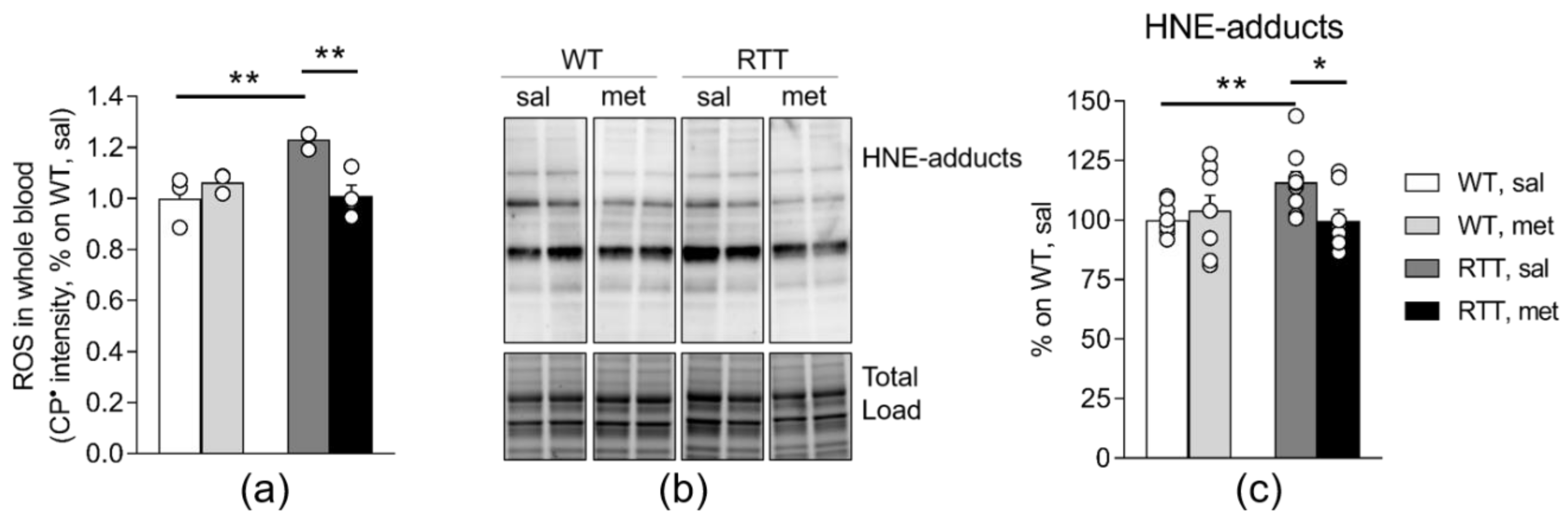
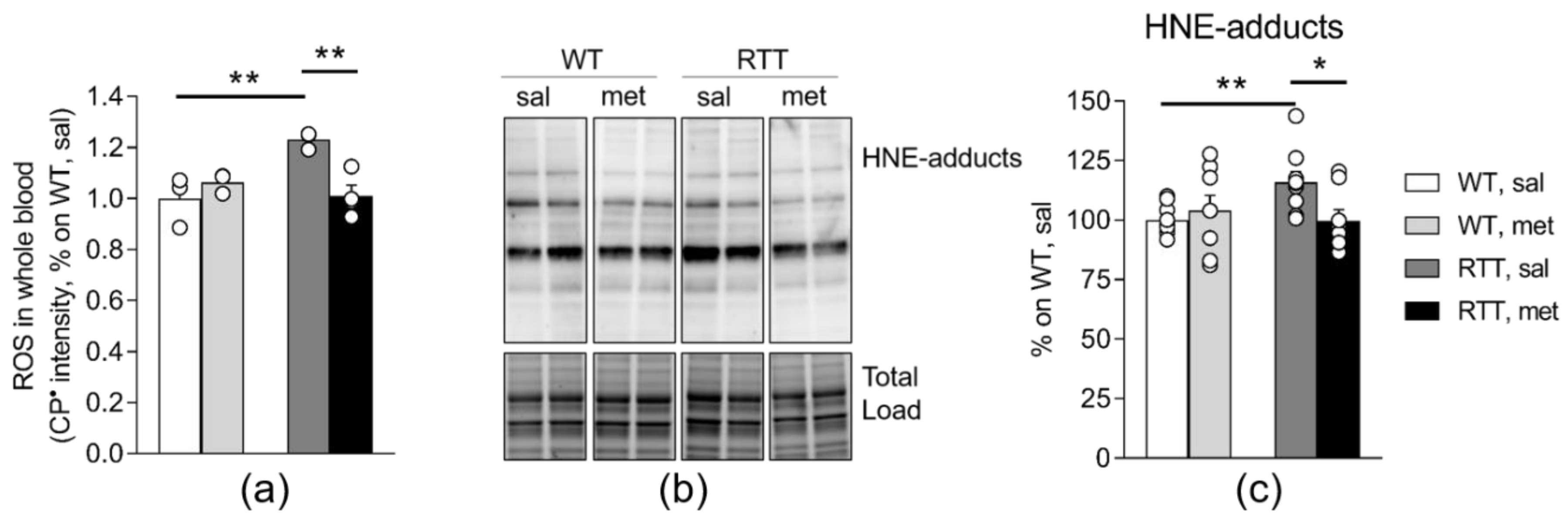
3.2. RTT Female Mice Show an Increased Oxidative Stress Status that Is Restored by Metformin both in the Brain and in the Blood
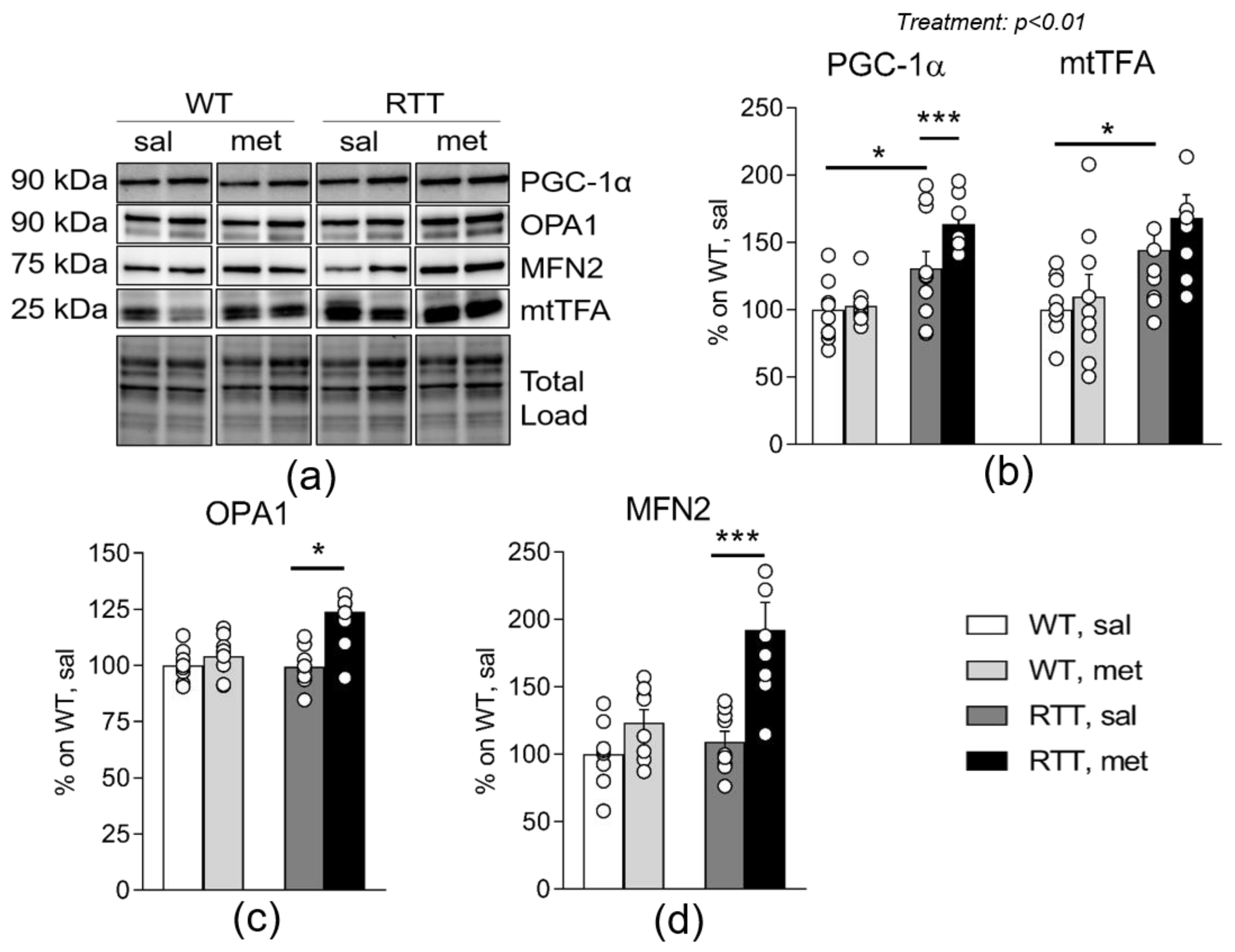
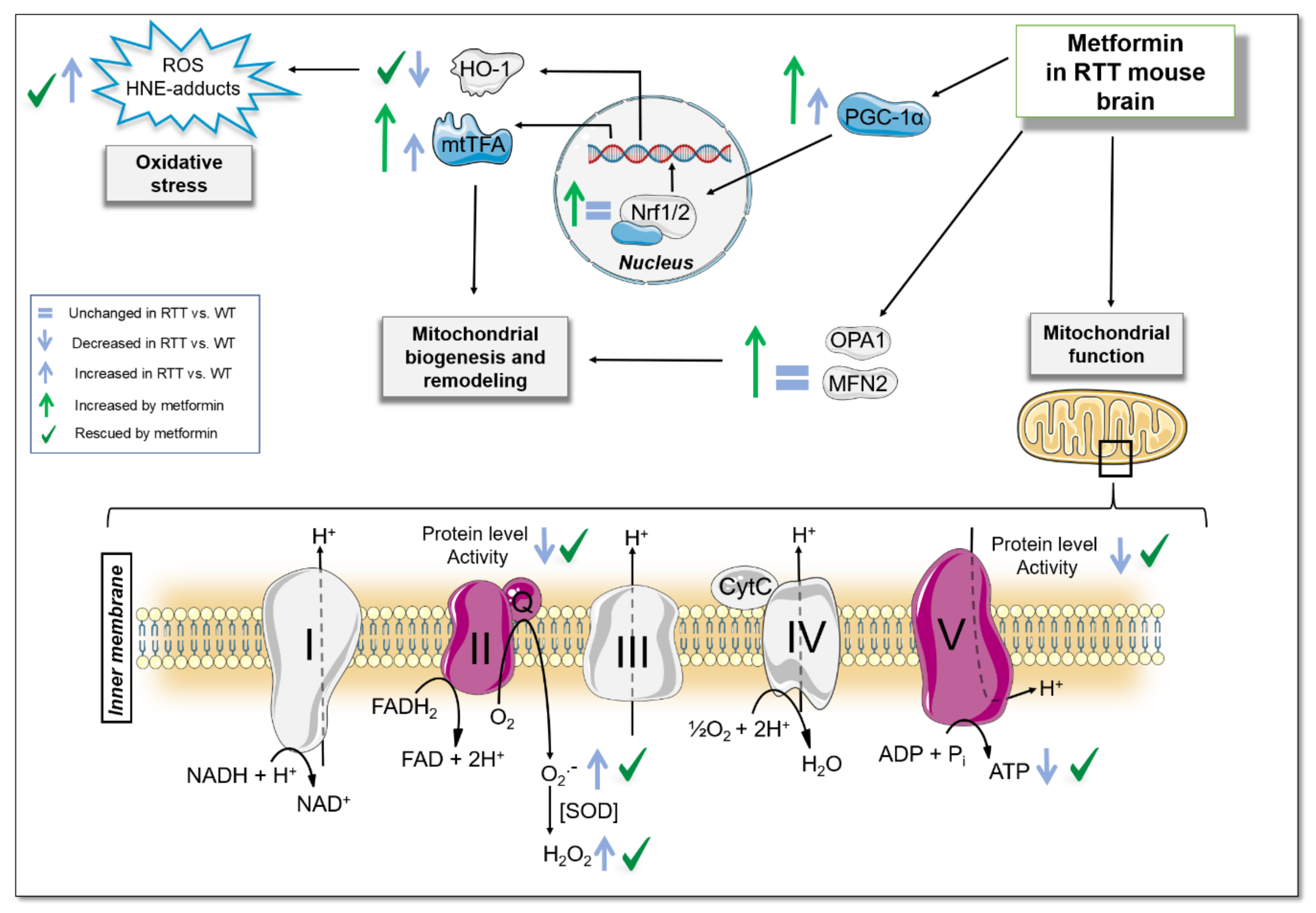
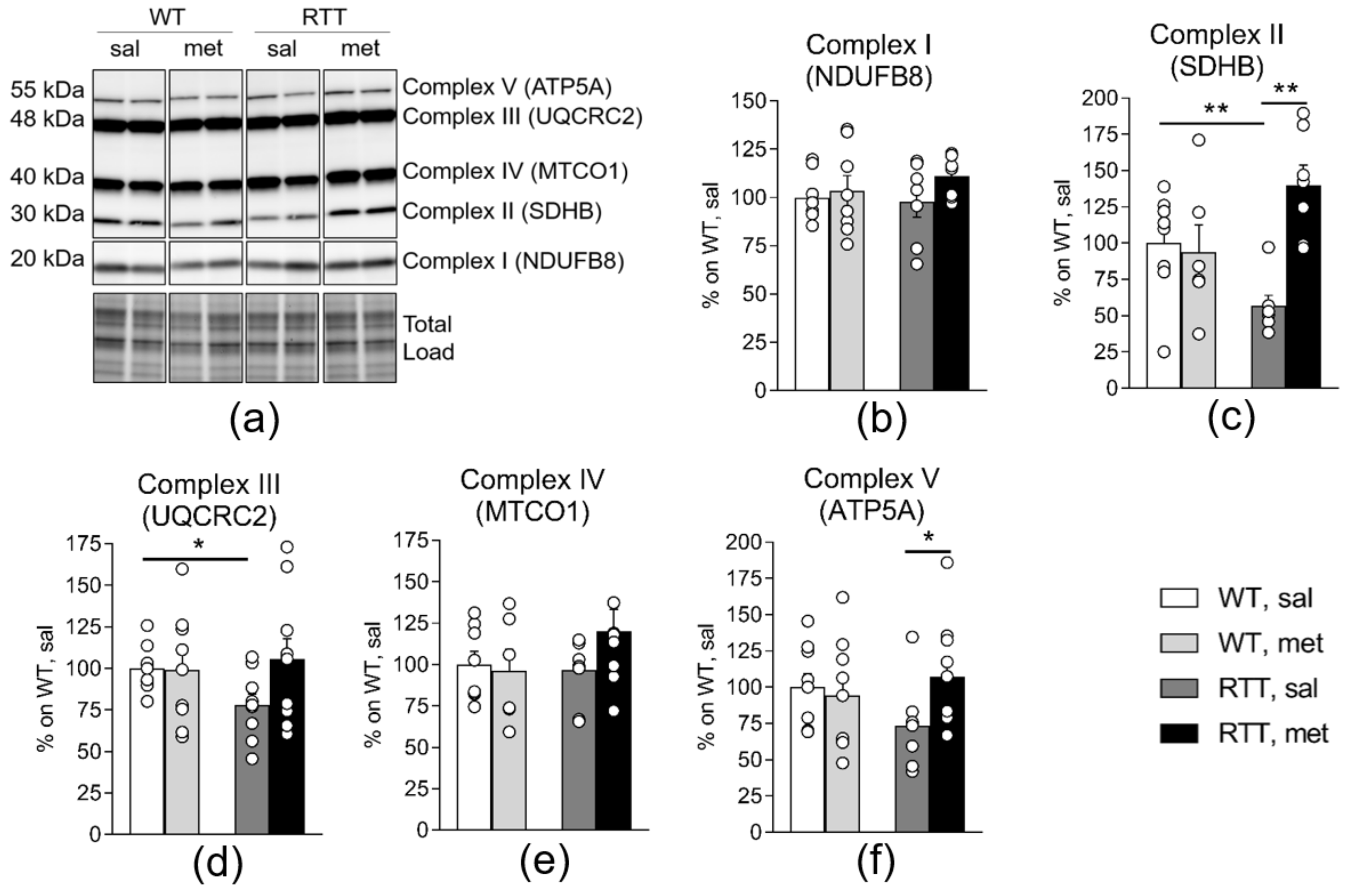
3.3. Metformin Systemic Treatment Boosts Pathways Related to Mitochondrial Biogenesis and Remodelling in the Brain of RTT Mice
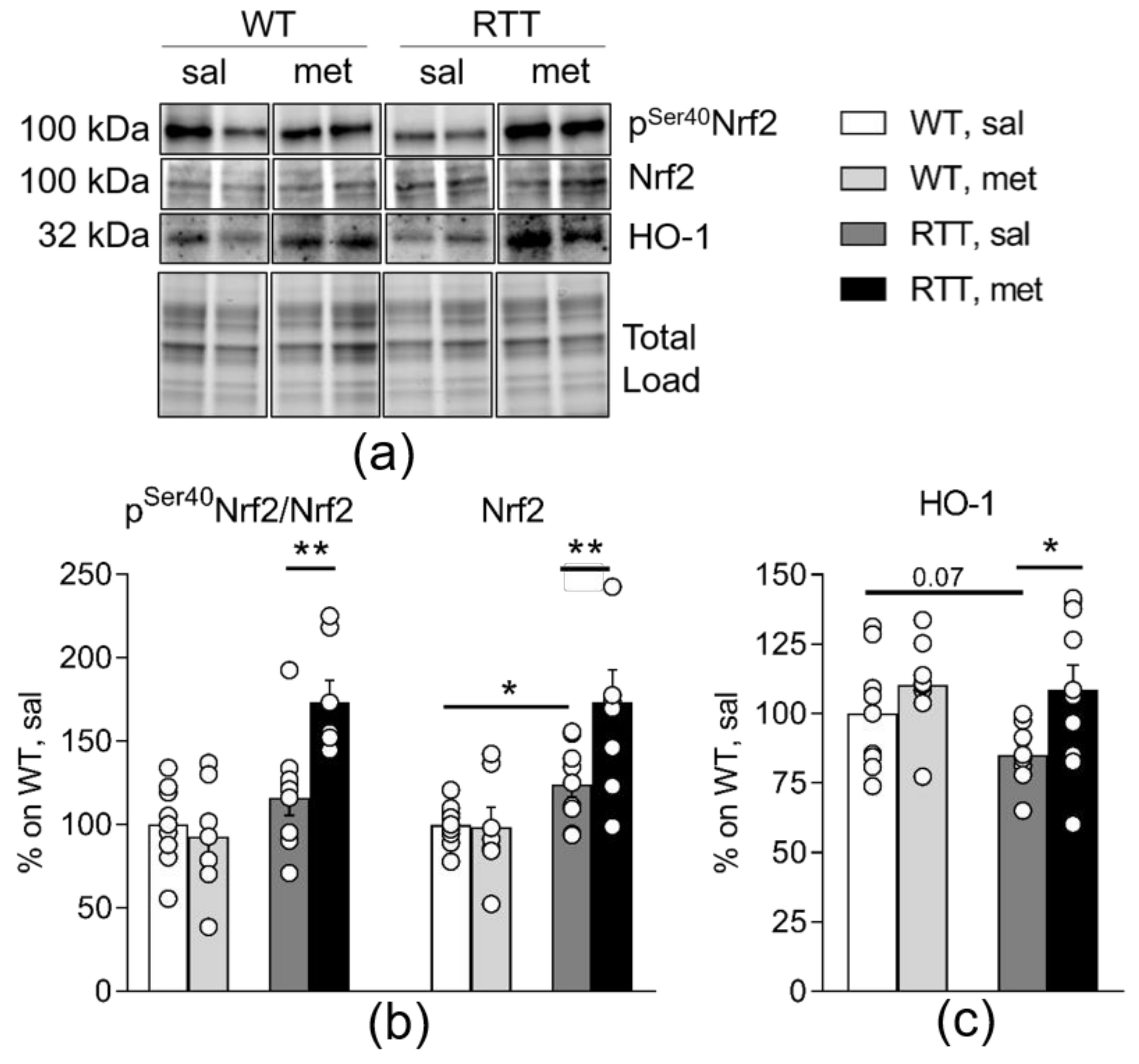
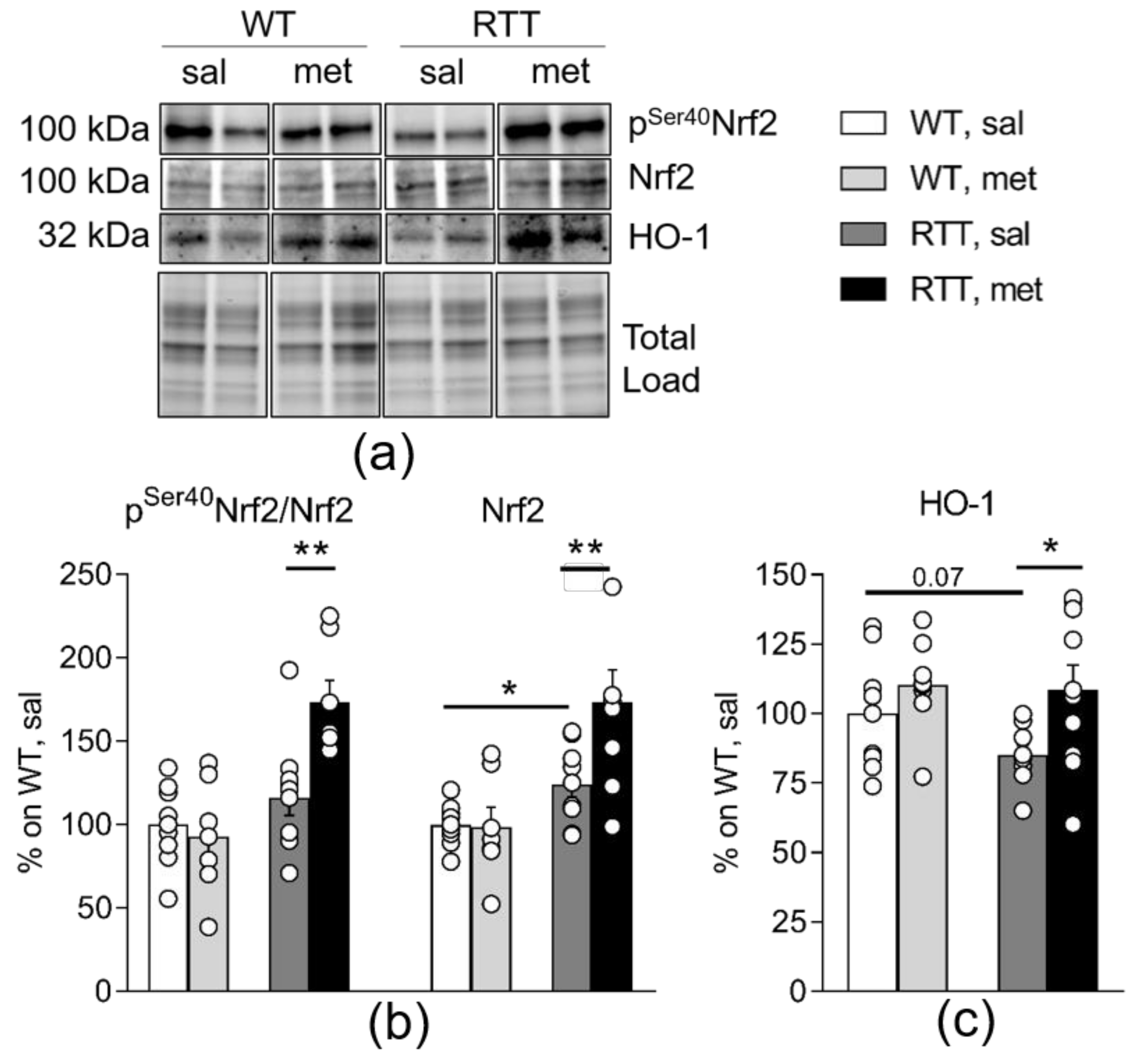
3.4. Metformin Systemic Treatment Boosts Pathways Related to the Antioxidant Response in the Brain of RTT Mice
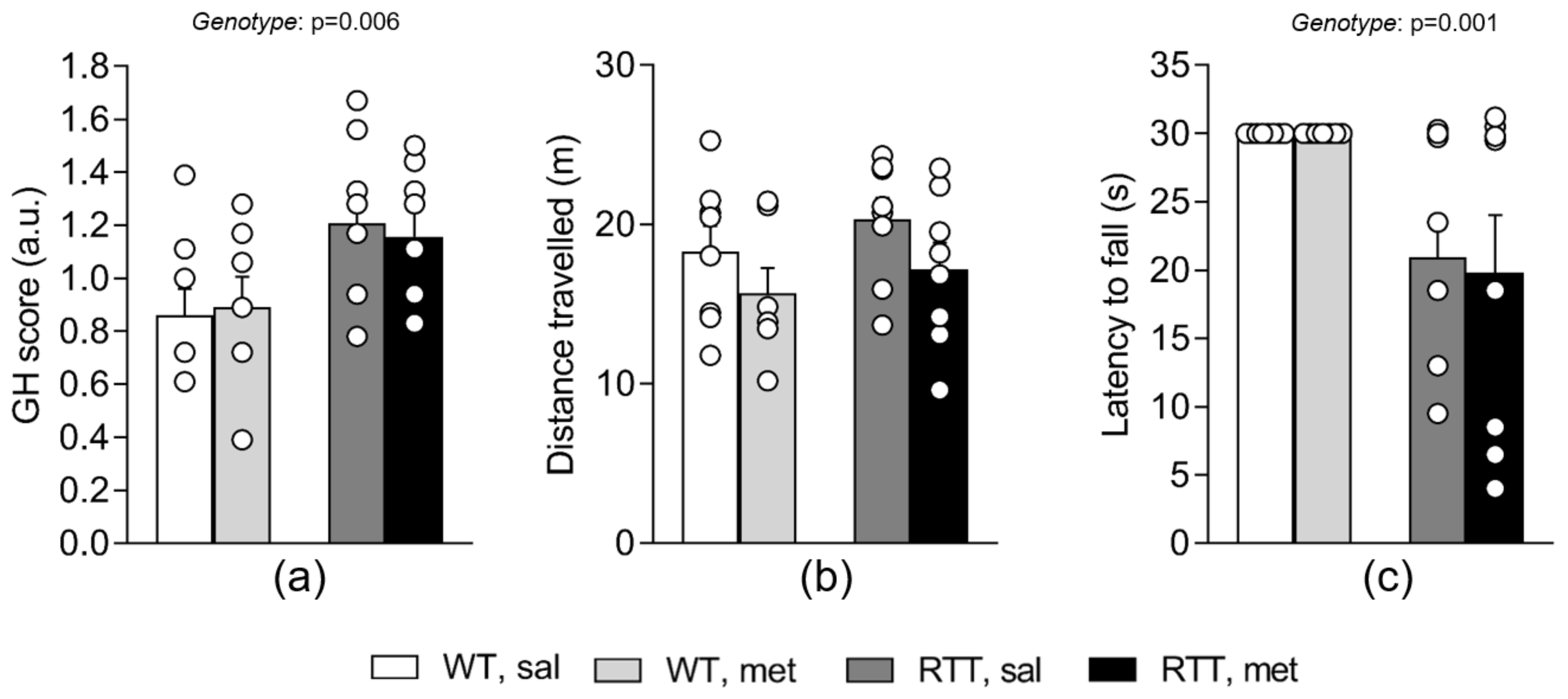
3.5. A 10-Day Long Treatment with Metformin at 100 mg/kg Dose Does Not Improve Behavioral Alterations in RTT Female Mice at an Advanced Stage of the Disease
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Alcocer-Gomez, E.; Garrido-Maraver, J.; Bullón, P.; Marín-Aguilar, F.; Cotán, D.; Carrión, A.M.; Alvarez-Suarez, J.M.; Giampieri, F.; Sánchez-Alcazar, J.A.; Battino, M.; et al. Metformin and caloric restriction induce an AMPK-dependent restoration of mitochondrial dysfunction in fibroblasts from Fibromyalgia patients. Biochim. Biophys. Acta 2015, 1852, 1257–1267. [Google Scholar] [CrossRef]
- Auger, C.; Sivayoganathan, T.; Abdullahi, A.; Parousis, A.; Pang, B.W.; Jeschke, M.G. Metformin adapts its cellular effects to bioenergetic status in a model of metabolic dysfunction. Sci. Rep. 2018, 8, 5646. [Google Scholar] [CrossRef]
- Kane, D.A.; Anderson, E.J.; Price, J.W., 3rd; Woodlief, T.L.; Lin, C.T.; Bikman, B.T.; Cortright, R.N.; Neufer, P.D. Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats. Free Radic. Biol. Med. 2010, 49, 1082–1087. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 2019, 4, e130681. [Google Scholar] [CrossRef]
- Pecinova, A.; Brázdová, A.; Drahota, Z.; Houštěk, J.; Mráček, T. Mitochondrial targets of metformin—Are they physiologically relevant? Biofactors 2019, 45, 703–711. [Google Scholar] [CrossRef]
- Vial, G.; Detaille, D.; Guigas, B. Role of mitochondria in the mechanism(s) of action of metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, E. Metformin-induced mitochondrial complex i inhibition: Facts, uncertainties, and consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Aatsinki, S.M.; Buler, M.; Salomäki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1alpha expression and selectively affects hepatic PGC-1alpha functions. Br. J. Pharmacol. 2014, 171, 2351–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashabi, G.; Khodagholi, F.; Khalaj, L.; Goudarzvand, M.; Nasiri, M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: Interference of AMPK/PGC-1alpha pathway. Metab. Brain. Dis. 2014, 29, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef]
- Gold, W.A.; Williamson, S.L.; Kaur, S.; Hargreaves, I.P.; Land, J.M.; Pelka, G.J.; Tam, P.P.; Christodoulou, J. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): Implications for the disease phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; Vigli, D.; Lacivita, E.; Leopoldo, M.; Laviola, G.; Vacca, R.A.; De Filippis, B. Stimulation of the brain serotonin receptor 7 rescues mitochondrial dysfunction in female mice from two models of Rett syndrome. Neuropharmacology 2017, 121, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Filosa, S.; Pecorelli, A.; D’Esposito, M.; Valacchi, G.; Hajek, J. Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome. Free Radic. Biol. Med. 2015, 88, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Leoni, G.; Cervellati, F.; Canali, R.; Signorini, C.; Leoncini, S.; Cortelazzo, A.; De Felice, C.; Ciccoli, L.; Hayek, J.; et al. Genes related to mitochondrial functions, protein degradation, and chromatin folding are differentially expressed in lymphomonocytes of Rett syndrome patients. Mediators Inflamm. 2013, 2013, 137629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, C.; Ciccoli, L.; Leoncini, S.; Signorini, C.; Rossi, M.; Vannuccini, L.; Guazzi, G.; Latini, G.; Comporti, M.; Valacchi, G.; et al. Systemic oxidative stress in classic Rett syndrome. Free Radic. Biol. Med. 2009, 47, 440–448. [Google Scholar] [CrossRef]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabrì, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8, 170216. [Google Scholar] [CrossRef] [Green Version]
- Kyle, S.M.; Saha, P.K.; Brown, H.M.; Chan, L.C.; Justice, M.J. MeCP2 co-ordinates liver lipid metabolism with the NCoR1/HDAC3 corepressor complex. Hum. Mol. Genet. 2016, 25, 3029–3041. [Google Scholar] [CrossRef] [Green Version]
- Justice, M.J.; Buchovecky, C.M.; Kyle, S.M.; Djukic, A. A role for metabolism in Rett syndrome pathogenesis: New clinical findings and potential treatment targets. Rare Dis. 2013, 1, e27265. [Google Scholar] [CrossRef] [Green Version]
- Ricceri, L.; De Filippis, B.; Laviola, G. Mouse models of Rett syndrome: From behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav. Pharmacol. 2008, 19, 501–517. [Google Scholar] [CrossRef] [Green Version]
- Shahbazian, M.D.; Zoghbi, H.Y. Rett syndrome and MeCP2: Linking epigenetics and neuronal function. Am. J. Hum. Genet. 2002, 71, 1259–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, D.M.; Berger-Sweeney, J.E.; Eubanks, J.H.; Justice, M.J.; Neul, J.L.; Pozzo-Miller, L.; Blue, M.E.; Christian, D.; Crawley, J.N.; Giustetto, M.; et al. Preclinical research in Rett syndrome: Setting the foundation for translational success. Dis. Model Mech. 2012, 5, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Cosentino, L.; Laviola, G.; De Filippis, B. Genes and sex hormones interaction in neurodevelopmental disorders. Neurosci. Biobehav. Rev. 2016, 67, 9–24. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Chiodi, V.; Adriani, W.; Lacivita, E.; Mallozzi, C.; Leopoldo, M.; Domenici, M.R.; Fuso, A.; Laviola, G. Long-lasting beneficial effects of central serotonin receptor 7 stimulation in female mice modeling Rett syndrome. Front. Behav. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigli, D.; Cosentino, L.; Raggi, C.; Laviola, G.; Woolley-Roberts, M.; De Filippis, B. Chronic treatment with the phytocannabinoid Cannabidivarin (CBDV) rescues behavioural alterations and brain atrophy in a mouse model of Rett syndrome. Neuropharmacology 2018, 140, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Vigli, D.; Cosentino, L.; Raggi, C.; Laviola, G.; Woolley-Roberts, M.; De Filippis, B. Metformin produces anxiolytic-like effects in rats by facilitating GABAA receptor trafficking to membrane. Br. J. Pharmacol. 2019, 176, 297–316. [Google Scholar]
- Zhou, W.; Kavelaars, A.; Heijnen, C.J. Metformin Prevents Cisplatin-Induced Cognitive Impairment and Brain Damage in Mice. PLoS ONE 2016, 11, e0151890. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, B.; Ricceri, L.; Laviola, G. Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes Brain Behav. 2010, 9, 213–223. [Google Scholar] [CrossRef]
- De Filippis, B.; Ricceri, L.; Laviola, G. Pharmacological stimulation of the brain serotonin receptor 7 as a novel therapeutic approach for Rett syndrome. Neuropsychopharmacology 2014, 39, 2506–2518. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Ricceri, L.; Vacca, R.A. Preservation of mitochondrial functional integrity in mitochondria isolated from small cryopreserved mouse brain areas. Anal. Biochem. 2014, 444, 25–31. [Google Scholar] [CrossRef]
- Valenti, D.; Tullo, A.; Caratozzolo, M.F.; Merafina, R.S.; Scartezzini, P.; Marra, E.; Vacca, R.A. Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 2010, 431, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A. Bioluminometric assay of ATP in mouse brain: Determinant factors for enhanced test sensitivity. J. Biosci. 2003, 28, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Manente, G.A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: Involvement of the cAMP/PKA signalling pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straface, E.; Marchesi, A.; Gambardella, L.; Metere, A.; Tarissi de Jacobis, I.; Viora, M.; Giordani, L.; Villani, A.; Del Principe, D. Does oxidative stress play a critical role in cardiovascular complications of Kawasaki disease? Antioxid. Redox Signal. 2012, 17, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Butterfield, Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di Domenico, F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Grosser, E.; Hirt, U.; Janc, O.A.; Menzfeld, C.; Fischer, M.; Kempkes, B.; Vogelgesang, S.; Manzke, T.U.; Opitz, L.; Salinas-Riester, G.; et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis. 2012, 48, 102–114. [Google Scholar] [CrossRef]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial dysfunction in the pathogenesis of Rett syndrome: Implications for Mitochondria-targeted therapies. Front. Cell. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Teng, Y.; Zhang, X.; Lv, X.; Yin, Y. Metformin Alleviated Abeta—Induced Apoptosis via the suppression of JNK MAPK signaling pathway in cultured hippocampal neurons. Biomed. Res. Int. 2016, 2016, 1421430. [Google Scholar] [PubMed] [Green Version]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A future therapy for neurodegenerative diseases: Theme: Drug discovery, development and delivery in Alzheimer’s disease guest editor: Davide Brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of Rett syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef]
- Suwa, M.; Egashira, T.; Nakano, H.; Sasaki, H.; Kumagai, S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J. Appl. Physiol. (1985) 2006, 101, 1685–1692. [Google Scholar] [CrossRef] [Green Version]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Pecorelli, A.; Belmonte, G.; Meloni, I.; Cervellati, F.; Gardi, C.; Sticozzi, C.; De Felice, C.; Signorini, C.; Cortelazzo, A.; Leoncini, S.; et al. Alteration of serum lipid profile, SRB1 loss, and impaired Nrf2 activation in CDKL5 disorder. Free Radic. Biol. Med. 2015, 86, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1alpha, Inflammation, and oxidative stress: An integrative view in metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Slack, R.S. Mitochondrial dynamics in the regulation of neurogenesis: From development to the adult brain. Dev. Dyn. 2018, 247, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.L.; Arslanian, S.; Peterokova, V.A.; Park, J.S.; Tomlinson, M.J. Effect of metformin in pediatric patients with type 2 diabetes: A randomized controlled trial. Diabetes Care 2002, 25, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuliani, I.; Urbinati, C.; Valenti, D.; Quattrini, M.C.; Medici, V.; Cosentino, L.; Pietraforte, D.; Di Domenico, F.; Perluigi, M.; Vacca, R.A.; et al. The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome. J. Clin. Med. 2020, 9, 1669. https://doi.org/10.3390/jcm9061669
Zuliani I, Urbinati C, Valenti D, Quattrini MC, Medici V, Cosentino L, Pietraforte D, Di Domenico F, Perluigi M, Vacca RA, et al. The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome. Journal of Clinical Medicine. 2020; 9(6):1669. https://doi.org/10.3390/jcm9061669
Chicago/Turabian StyleZuliani, Ilaria, Chiara Urbinati, Daniela Valenti, Maria Cristina Quattrini, Vanessa Medici, Livia Cosentino, Donatella Pietraforte, Fabio Di Domenico, Marzia Perluigi, Rosa Anna Vacca, and et al. 2020. "The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome" Journal of Clinical Medicine 9, no. 6: 1669. https://doi.org/10.3390/jcm9061669
APA StyleZuliani, I., Urbinati, C., Valenti, D., Quattrini, M. C., Medici, V., Cosentino, L., Pietraforte, D., Di Domenico, F., Perluigi, M., Vacca, R. A., & De Filippis, B. (2020). The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome. Journal of Clinical Medicine, 9(6), 1669. https://doi.org/10.3390/jcm9061669