Evaluation of Innate Immune Mediators Related to Respiratory Viruses in the Lung of Stable COPD Patients


 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Methods
2.1. Subjects
2.2. Lung Function Tests and Volumes
2.3. Fiberoptic Bronchoscopy, Collection, and Processing of Bronchial Biopsies
2.4. Collection and Processing of the Peripheral Lung Tissue
2.5. Immunohistochemistry Analysis on OCT-embedded Bronchial Biopsies and Peripheral Lung Tissue
2.6. Scoring System and Quantification of Immunohistochemistry in the Bronchial Biopsies
2.7. Scoring System for Immunohistochemistry in the Peripheral Lung Tissue
2.8. Collection and Processing of Broncho-Alveolar Lavage
2.9. Extraction of RNA/DNA and qRT-PCR for Viral Load Quantification in Bronchial Rings and Lung Parenchyma
2.10. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Subjects
3.2. Measurement of Inflammatory Cells in the Bronchial Biopsies of COPD Patients
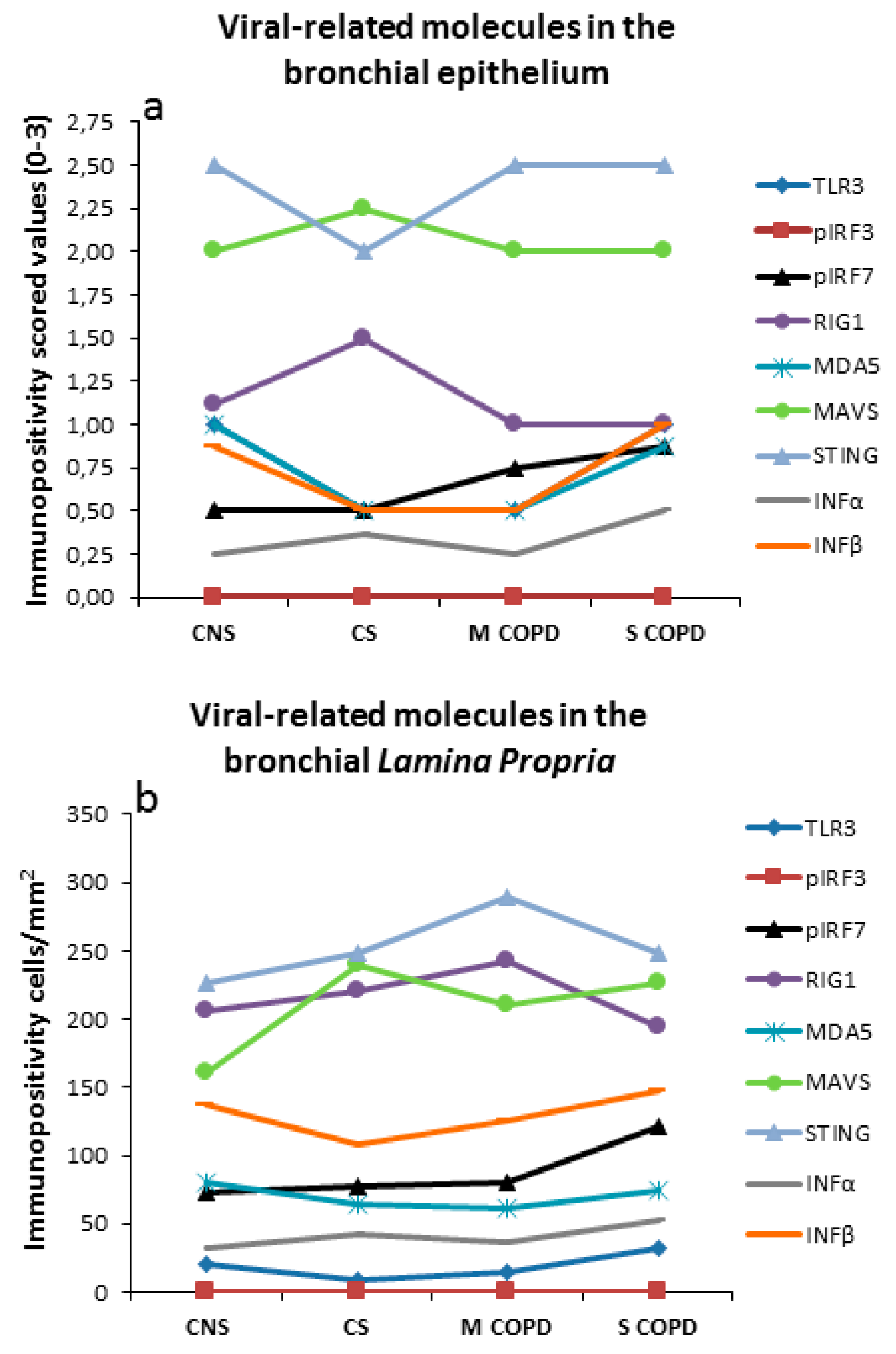
3.3. Immunohistochemistry of Innate Immune Mediators Related to Respiratory Viruses in Bronchial Biopsies
3.4. Immunohistochemistry in Bronchial Epithelium
3.5. Immunohistochemistry in Bronchial Lamina Propria
3.6. Immunohistochemistry of Innate Immune Mediators Related to Respiratory Viruses in the Peripheral Lung Tissue
3.7. ELISA Assays of Innate Immune Mediators Related to Respiratory Viruses in the BAL Supernatants
3.8. Correlations among Innate Immune Mediators Related to Respiratory Viruses, Clinical Parameters, and Inflammatory Cells in Bronchial Biopsies
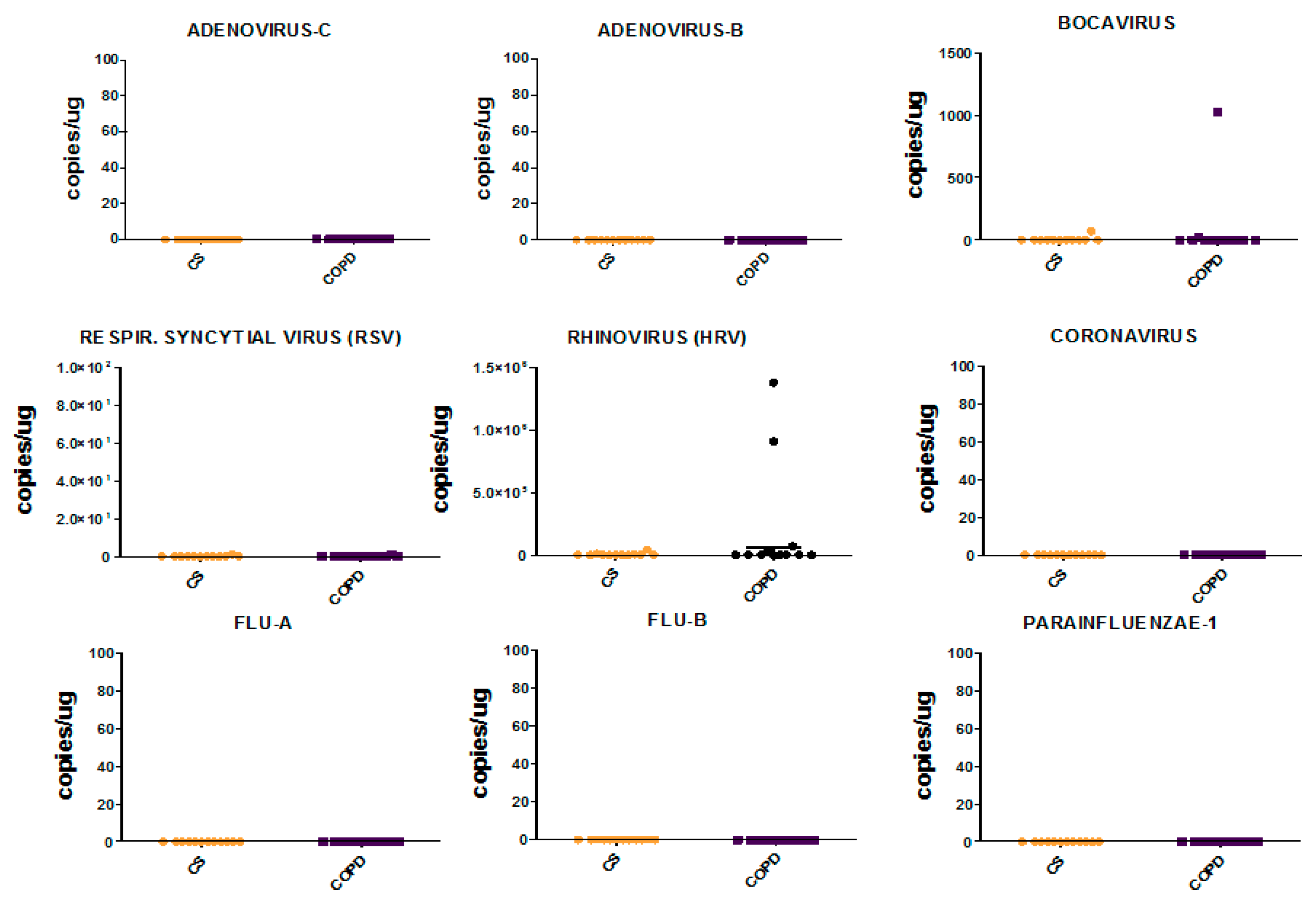
3.9. Viral Load in Bronchial Biopsies
3.10. Viral Load in Bronchial Rings and Peripheral Lung Tissue
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnes, P.J. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin. Chest Med. 2014, 35, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Ricciardolo, F.L.; Capelli, A.; Adcock, I.M.; Donner, C.F. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: An overview. Clin. Exp. Allergy 2004, 34, 1156–1167. [Google Scholar] [CrossRef]
- Caramori, G.; Casolari, P.; Barczyk, A.; Durham, A.L.; Di Stefano, A.; Adcock, I. COPD immunopathology. In Semin Immunopathol; Springer: Berlin/Heidelberg, Germany, 2016; Volume 38, pp. 497–515. [Google Scholar]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; e Sousa, C.R. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9–mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Génin, P.; Vaccaro, A.; Civas, A. The role of differential expression of human interferon—A genes in antiviral immunity. Cytokine Growth Factor Rev. 2009, 20, 283–295. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T.; et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, K.; Sugiyama, T.; Matsumoto, M.; Tanaka, T.; Saito, M.; Hemmi, H.; Ohara, O.; Akira, S.; Kaisho, T. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 2006, 440, 949–953. [Google Scholar] [CrossRef]
- Sharma, S. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M.; Akira, S., Jr.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [Green Version]
- Malur, M.; Gale, M.; Krug, R.M., Jr. LGP2 downregulates interferon production during infection with seasonal human influenza A viruses that activate interferon regulatory factor 3. J. Virol. 2012, 86, 10733–10738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, K.S.; Randall, R.E.; Goodbourn, S. LGP2 plays a critical role in sensitizing Mda-5 to activation by double-stranded RNA. PLoS ONE 2013, 8, e64202. [Google Scholar] [CrossRef] [Green Version]
- Nakhaei, P.; Hiscott, J.; Lin, R. STING-ing the antiviral pathway. J. Mol. Cell Biol. 2009, 2, 110–112. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Vilaysane, A.; Muruve, D.A. The innate immune response to DNA. Semin. Immunol. 2009, 21, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Chen, G.; Korfhagen, T.R.; Karp, C.; Impey, S.; Xu, Y.; Randell, S.H.; Kitzmiller, J.; Maeda, Y.; Haitchi, H.M.; Sridharan, A.; et al. Foxa3 induces goblet cell metaplasia and inhibits innate antiviral immunity. Am. J. Respir. Crit. Care Med. 2014, 189, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Caramori, G.; Barczyk, A.; Vicari, C.; Brun, P.; Zanini, A.; Cappello, F.; Garofano, E.; Padovani, A.; Contoli, M.; et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax 2014, 69, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Ricciardolo, F.L.; Caramori, G.; Adcock, I.M.; Chung, K.F.; Barnes, P.J.; Brun, P.; Leonardi, A.; Ando, F.; Vallese, D.; et al. Bronchial inflammation and bacterial load in stable COPD is associated with TLR4 overexpression. Eur. Respir. J. 2017, 49, 1602006. [Google Scholar] [CrossRef] [Green Version]
- Sze, M.A.; Dimitriu, P.A.; Suzuki, M.; McDonough, J.E.; Campbell, J.D.; Brothers, J.F.; Erb-Downward, J.R.; Huffnagle, G.B.; Hayashi, S.; Elliott, W.M.; et al. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2015, 192, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, R.P.; Erb-Downward, J.R.; Huffnagle, G.B. Homeostasis and its disruption in the lung microbiome. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1047–L1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visseaux, B.; Burdet, C.; Voiriot, G.; Lescure, F.-X.; Chougar, T.; Brugière, O.; Crestani, B.; Casalino, E.; Charpentier, C.; Descamps, D.; et al. Prevalence of respiratory viruses among adults, by season, age, respiratory tract region and type of medical unit in Paris, France, from 2011 to 2016. PLoS ONE 2017, 12, e0180888. [Google Scholar] [CrossRef] [Green Version]
- Kilburn, K.H. Functional morphology of the distal lung. In International Review of Cytology; Academic Press: Cambridge, MA, USA, 1974; Volume 37, pp. 153–270. [Google Scholar]
- Wiebe, B.M.; Laursen, H. Lung morphometry by unbiased methods in emphysema: Bronchial and blood vessel volume, alveolar surface area and capillary length. Apmis 1998, 106, 651–656. [Google Scholar] [CrossRef]
- Wilkinson, T.M.A.; Donaldson, G.C.; Johnston, S.L.; Openshaw, P.J.; Wedzicha, J.A. Respiratory syncytial virus, airway inflammation, and FEV1Decline in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 871–876. [Google Scholar] [CrossRef]
- Seemungal, T.; Harper-Owen, R.; Bhowmik, A.; Morić, I.; Sanderson, G.; Message, S.; Maccallum, P.; Meade, T.W.; Jeffries, D.J.; Johnston, S.L.; et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 164, 1618–1623. [Google Scholar] [CrossRef]
- Wilkinson, T.M.A.; Aris, E.; Bourne, S.; Clarke, S.C.; Peeters, M.; Pascal, T.G.; Schoonbroodt, S.; Tuck, A.C.; Kim, V.; Ostridge, K.K.; et al. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax 2017, 72, 919–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, T.E.; Marley, A.-M.; Baxter, N.; Christie, S.N.; O’Neill, H.J.; Elborn, J.S.; Coyle, P.; Kidney, J.C. Respiratory viral infection in exacerbations of COPD. Respir. Med. 2008, 102, 1575–1580. [Google Scholar] [CrossRef] [Green Version]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef]
- Hurst, J.R.; Vestbo, J.; Anzueto, A.; Locantore, N.; Müllerova, H.; Tal-Singer, R.; Miller, B.; Lomas, D.A.; Agusti, A.; MacNee, W.; et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N. Engl. J. Med. 2010, 363, 1128–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utokaparch, S.; Sze, M.A.; Gosselink, J.V.; McDonough, J.E.; Elliott, W.M.; Hogg, J.C.; Hegele, R.G. Respiratory viral detection and small airway inflammation in lung tissue of patients with stable, mild COPD. COPD J. Chronic Obstr. Pulm. Dis. 2013, 11, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Chiatti, C.; Barbadoro, P.; Marigliano, A.; Ricciardi, A.; Di Stanislao, F.; Prospero, E. Determinants of influenza vaccination among the adult and older Italian population with chronic obstructive pulmonary disease: A secondary analysis of the multipurpose ISTAT Survey on Health and Health Care use. Hum. Vaccines 2011, 7, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Formica, M.A.; Hennessey, P.A.; Criddle, M.M.; Sullender, W.M.; Walsh, E.E. Detection of respiratory syncytial virus in adults with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 639–643. [Google Scholar] [CrossRef] [Green Version]
- McManus, T.E.; Marley, A.-M.; Baxter, N.; Christie, S.N.; Elborn, J.S.; Heaney, L.G.; Coyle, P.; Kidney, J.C. Acute and latent adenovirus in COPD. Respir. Med. 2007, 101, 2084–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, A.; Walsh, E.E.; Formica, M.A.; Hennessey, P.A.; Criddle, M.M.; Peterson, D.R.; Baran, A.; Falsey, A.R. Factors associated with symptomatic rhinovirus infection in patients with COPD. J. Clin. Virol. 2012, 55, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Quint, J.K.; Donaldson, G.C.; Goldring, J.J.; Baghai-Ravary, R.; Hurst, J.R.; Wedzicha, J.A. Serum IP-10 as a biomarker of human rhinovirus infection at exacerbation of COPD. Chest 2009, 137, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.L.; Turner, R.B.; Braciale, T.; Heymann, P.W.; Borish, L. Pathogenesis of rhinovirus infection. Curr. Opin. Virol. 2012, 2, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Birger, R.; Morita, H.; Comito, D.; Filip, I.; Galanti, M.; Lane, B.; Ligon, C.; Rosenbloom, D.; Shittu, A.; Ud-Dean, M.; et al. Asymptomatic shedding of respiratory virus among an ambulatory population across seasons. mSphere 2018, 3, e00249-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Groups | n | Age (years) | M/F | Pack Years | Ex/Current Smokers | FEV1 (% pred.) pre-β2 | FEV1 (% pred.) post-β2 | FEV1/FVC (%) |
|---|---|---|---|---|---|---|---|---|
| Control non-smokers | 12 | 63 ± 13 | 10/2 | 0 | 0 | 117 ± 18 | ND | 86 ± 10 |
| Control smokers with normal lung function | 12 | 61 ± 7 | 9/3 | 43 ± 26 | 2/10 | 104 ± 13 | ND | 81 ± 6 |
| COPD stages I and II (mild/moderate) | 16 | 71 ± 8 § | 14/3 | 50 ± 28 | 6/11 | 63 ± 11 # | 67 ± 13 | 57 ± 9 # |
| COPD stages III and IV (severe/very severe) | 18 | 66 ± 9 § | 11/7 | 54 ± 36 | 13/5 | 35 ± 8 #,& | 38 ± 9 | 44 ± 10 #,& |
| Groups | n | Age (years) | Sex M/F | Ex/Current Smokers | Pack-Years | Chronic Bronchitis (Yes/No) | FEV1 (% pred.) | FEV1/FVC% |
|---|---|---|---|---|---|---|---|---|
| Control smokers | 12 | 70.1 ± 2 | 11/1 | 6/6 | 33.6 ± 9.7 | no | 101.8 ± 3.8 | 74.0 ± 2.0 |
| COPD patients | 12 | 68.1 ± 2 | 6/6 | 10/2 | 44.4 ± 24.4 | no | 86.9 ± 4.8 * | 65.7 ± 2.6 & |
| Epithelium (Score 0–3) | Control Non-Smokers | Control Smokers | Mild/Moderate COPD | Severe/Very Severe COPD | Kruskal Wallis p Value |
|---|---|---|---|---|---|
| TLR3 | 1.0 (0.12–1.5) | 0.5 (0.12–1.5) | 0.5 (0.12–1.25) | 1.0 (0.12–2.5) & | 0.063 |
| TLR7 | 0.75 (0.25–1) | 0.75 (0.5–1.25) | 0.5 (0.25–1) | 0.75 (0.25–1.25) | 0.373 |
| TLR8 | 0.25 (0–0.5) | 0.25 (0.12–0.5) | 0.25 (0–0.75) | 0.25 (0.12–1) | 0.499 |
| TLR9 | 0.5 (0.12–0.75) | 0.37 (0.12–1) | 0.5 (0–1) | 0.5 (0.12–1.5) | 0.851 |
| TICAM (TRIF) | 2 (1.5–2.5) | 2.12 (1.5–2.75) | 2 (0.75–2.5) | 2 (0.75–2.75) | 0.456 |
| IRF3 | 0.25 (0–0.25) | 0.25 (0–0.25) | 0.25 (0–1) | 0.25 (0–0.5) | 0.406 |
| Phospho-IRF3 | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) | n.d. |
| IRF7 | 1.5 (0.75–1.75) | 0.75 (0.25–1.75) * | 0.5 (0.25–1.5) * | 0.75 (0.25–2.5) | 0.077 |
| Phospho-IRF7 | 0.5 (0.25–1) | 0.5 (0–1.5) | 0.75 (0.25–1.25) | 0.87 (0.5–1) * | 0.214 |
| DDX58 (RIG1) | 1.12 (0.5–2.5) | 1.5 (0.5–2) | 1 (0.5–2) | 1 (0.5–2.5) | 0.996 |
| MDA5 | 1 (0.5–1.5) | 0.5 (0.5–1.5) | 0.5 (0.25–1.5) * | 0.87 (0.25–1.5) | 0.064 |
| DHX58 (LGP2) | 2 (1.5–2.5) | 1.75 (0.75–2.5) | 1.75 (1–2.5) | 1.62 (1–2.5) | 0.402 |
| MAVS | 2 (1.25–2.5) | 2.25 (1.25–3) | 2 (1–3) | 2 (1–3) | 0.472 |
| STING (TMEM173) | 2.5 (1.5–2.5) | 2 (1.5–2.5) | 2.5 (1.5–3) | 2.5 (1.5–3) | 0.208 |
| DAI (ZBP1) | 2 (1–2.5) | 2.5 (1.5–3) | 2 (1–2.5) | 2.5 (1.5–3) | 0.197 |
| FOXA3 | 1.75 (0.5–2) | 1.37 (0.5–1.5) | 1.5 (0.75–2) | 1.5 (0.5–3) | 0.797 |
| IFNα | 0.25 (0–0.75) | 0.37 (0–1) | 0.25 (0.25–1) | 0.5 (0.25–1.25) | 0.169 |
| IFNβ | 0.87 (0.5–1.5) | 0.5 (0.25–1.5) | 0.5 (0.25–1) * | 1 (0.5–1.5) § | 0.061 |
| Lamina Propria (cells/mm2) | |||||
| TLR3 | 21 (4–64) | 9 (0–48) | 14 (4–85) | 32 (0–263) & | 0.128 |
| TLR7 | 99 (5–161) | 95 (70–148) | 120 (16–242) | 123 (26–229) | 0.557 |
| TLR8 | 0 (0–43) | 0 (0–5) | 4 (0–41) | 3 (0–21) & | 0.108 |
| TLR9 | 4 (0–52) | 5 (0–15) | 13 (0–53) & | 4 (0–90) | 0.211 |
| TICAM (TRIF) | 183 (117–348) | 219 (145–312) | 206 (71–425) | 227 (129–387) | 0.923 |
| IRF3 | 17 (0–39) | 13 (4–92) | 24 (0–148) | 13 (0–64) | 0.541 |
| Phospho-IRF3 | 0 (0–0) | 0 (0–0) | 0 (0–4) | 0 (0–8) | 0.594 |
| IRF7 | 92 (45–193) | 79 (28–210) | 74 (24–206) | 87 (32–348) | 0.875 |
| Phospho-IRF7 | 73 (40–204) | 77 (0–203) | 81 (32–272) | 121 (40–366) | 0.444 |
| DDX58 (RIG1) | 206 (118–274) | 220 (77–272) | 243 (168-403) | 194 (114–281) | 0.247 |
| MDA5 | 81 (32–164) | 65 (29–187) | 62 (24–161) | 75 (32–186) | 0.908 |
| DHX58 (LGP2) | 303 (140–446) | 335 (174–554) | 343 (193–705) | 330 (148–559) | 0.897 |
| MAVS | 161 (51–226) | 239 (55–426) * | 210 (89–371) * | 226 (60–521) | 0.136 |
| STING (TMEM173) | 226 (86–400) | 248 (51–460) | 289 (97–435) | 249 (161–511) | 0.586 |
| DAI (ZBP1) | 202 (161–328) | 254 (203–387) | 290 (26–488) | 267 (153–405) | 0.237 |
| FOXA3 | 188 (73–343) | 173 (81–294) | 135 (97–333) | 175 (82–277) | 0.650 |
| IFNα | 32 (9–118) | 42 (6–87) | 37 (8–312) | 53 (11–161) | 0.447 |
| IFNβ | 137 (74–258) | 108 (43–276) | 126 (52–290) | 148 (78–287) | 0.187 |
| CD4 | 164 (101–212) | 246 (37–500) | 258 (107–731) | 252 (66–470) | 0.206 |
| CD8 | 147 (76–301) | 179 (86–657) | 195 (86–523) | 244 (111–355) * | 0.365 |
| CD68 | 284 (128–516) | 275 (97–904) | 367 (158–759) | 340 (204–1054) | 0.671 |
| Neutrophil Elastase | 93 (58–166) | 97 (45–308) | 94 (28–512) | 151 (47–470) *,& | 0.045 |
| Localization | Control Smokers with Normal Lung Function | COPD Patients | Mann-Witney U-Test p Value |
|---|---|---|---|
| Bronchiolar epithelium (score 0–3) | |||
| RIG1 | 0.70 (0.78) | 0.58 (0.36) | 0.185 |
| MDA5 | 0.96 (0.85) | 0.97 (0.45) | 0.689 |
| LGP2 | 0.12 (0.12) | 0.12 (0.05) | 0.597 |
| MAVS | 1.4 (0.68) | 0.75 (0.71) | 0.224 |
| STING | 1.60 (0.50) | 1.10 (0.77) | 0.037 |
| DAI | 0.25 (0.05) | 0.12 (0.02) | 0.146 |
| FOXA3 | 0.68 (0.81) | 0.75 (0.46) | 0.901 |
| IFNα | 0.30 (0.30) | 0.42 (0.31) | 0.435 |
| IFNβ | 0.25 (0.12) | 0.33 (0.11) | 0.123 |
| Bronchiolar submucosa (score 0–3) | |||
| RIG1 | 0.47 (0.44) | 0.46 (0.25) | 0.267 |
| MDA5 | 0.50 (0.33) | 0.50 (0.19) | 0.666 |
| LGP2 | 0.10 (0.10) | 0.12 (0.00) | 0.641 |
| MAVS | 0.5 (0.0) | 0.25 (0.25) | 0.068 |
| STING | 1.10 (0.25) | 0.90 (0.50) | 0.114 |
| DAI | 0.12 (0.05) | 0.10 (0.32) | 0.482 |
| FOXA3 | 0.50 (0.25) | 0.50 (0.12) | 0.982 |
| IFNα | 0.38 (0.27) | 0.43 (0.19) | 0.544 |
| IFNβ | 0.11 (0.17) | 0.17 (0.14) | 0.538 |
| Alveolar septa(score 0–3) | |||
| RIG1 | 0.42 (0.23) | 0.42 (0.24) | 0.460 |
| MDA5 | 0.50 (0.04) | 0.50 (0.03) | 0.758 |
| LGP2 | 1.00 (0.50) | 1.5 (0.75) | 0.112 |
| MAVS | 0.5 (0.18) | 0.62 (0.25) | 0.324 |
| STING | 0.50 (0.25) | 0.25 (0.00) | 0.057 |
| DAI | 0.25 (0.25) | 0.25 (0.05) | 0.218 |
| FOXA3 | 0.50 (0.05) | 0.50 (0.02) | 0.704 |
| IFNα | 0.40 (0.19) | 0.43 (0.16) | 0.885 |
| IFNβ | 0.25 (0.16) | 0.25 (0.13) | 0.644 |
| Alveolar macrophages (score 0–3) | |||
| RIG1 | 1.35 (1.12) | 1.14 (0.42) | 0.340 |
| MDA5 | 1.14 (0.75) | 1.46 (0.57) | 0.460 |
| LGP2 | 1.50 (0.25) | 2.00 (0.75) | 0.488 |
| MAVS | 0.5 (0.43) | 0.62 (0.37) | 0.853 |
| STING | 2.50 (0.50) | 2.00 (1.00) | 0.126 |
| DAI | 0.75 (0.93) | 0.50 (0.50) | 0.157 |
| FOXA3 | 0.50 (0.31) | 0.50 (0.43) | 0.939 |
| IFNα | 1.40 (0.44) | 1.33 (0.62) | 0.839 |
| IFNβ | 0.62 (0.28) | 0.80 (0.19) | 0.084 |
| Control Non-Smokers | Control Smokers with Normal Lung Function | Mild to Moderate COPD | |
|---|---|---|---|
| Number | 8 | 9 | 12 |
| Age (years) | 66.9 ± 2.9 | 60.7 ± 3.3 | 71.7 ± 1.6 * |
| Sex (M/F) | 1/7 | 9/0 | 9/3 |
| Ex/current smokers | -- | 4/5 | 8/4 |
| Pack-years | 0 | 40.1 ± 6.6 | 60.6 ± 13.6 |
| Chronic bronchitis | 2 | 5 | 5 |
| FEV1% predicted | 106.5 ± 3.2 | 97.6 ± 2.8 | 59.9 ± 5.1 & |
| FEV1/FVC% | 81.9 ± 2.5 | 83.5 ± 2.8 | 54.8 ± 2.7 & |
| Authors | Seemungal | Wilkinson 2006 | Wilkinson 2017 | Utokapark | Mc Manus 2007 | Mc Manus 2008 | Papi | Falsey |
|---|---|---|---|---|---|---|---|---|
| Weeks from last exacerbation | 4 | 4 | 4 | 2 | 8 | 8 | 8–10 | N.A |
| % patients treated with steroids | 97 ICS | NA | NA | 10 ICS | NA | 100 ICS 9% Oral | 97 ICS | 67 ICS 20 Oral |
| Steroids/daily mean, Beclomethasone equivalent dosage | 1120 | 1000 | NA | NA | NA | 948 | 980 | NA |
| Oral steroids | No | No | No | NA | NA | Yes (9%) | No | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Anna, S.E.; Maniscalco, M.; Carriero, V.; Gnemmi, I.; Caramori, G.; Nucera, F.; Righi, L.; Brun, P.; Balbi, B.; Adcock, I.M.; et al. Evaluation of Innate Immune Mediators Related to Respiratory Viruses in the Lung of Stable COPD Patients. J. Clin. Med. 2020, 9, 1807. https://doi.org/10.3390/jcm9061807
D’Anna SE, Maniscalco M, Carriero V, Gnemmi I, Caramori G, Nucera F, Righi L, Brun P, Balbi B, Adcock IM, et al. Evaluation of Innate Immune Mediators Related to Respiratory Viruses in the Lung of Stable COPD Patients. Journal of Clinical Medicine. 2020; 9(6):1807. https://doi.org/10.3390/jcm9061807
Chicago/Turabian StyleD’Anna, Silvestro E., Mauro Maniscalco, Vitina Carriero, Isabella Gnemmi, Gaetano Caramori, Francesco Nucera, Luisella Righi, Paola Brun, Bruno Balbi, Ian M Adcock, and et al. 2020. "Evaluation of Innate Immune Mediators Related to Respiratory Viruses in the Lung of Stable COPD Patients" Journal of Clinical Medicine 9, no. 6: 1807. https://doi.org/10.3390/jcm9061807