Ursodeoxycholic Acid Halts Pathological Neovascularization in a Mouse Model of Oxygen-Induced Retinopathy


 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Animal Model of Oxygen-Induced Retinopathy (OIR)
2.2. Bile Acids Treatment
2.3. Central Vaso-Obliteration and Neovascularization
2.4. Immunofluorescence Staining
2.5. Immunoblotting
2.6. Dot Blot Snalysis
2.7. Measurement of Retinal Vascular Leakage
2.8. Dihydroethidium Staining for Detection of Superoxide
2.9. Cytokines Assay
2.10. Cells and Angiogenesis Assay
2.11. In Vitro Fluorescein Isothiocyanate–Dextran (FITC-Dextran) Leakage Assay
2.12. Statistical Analysis
3. Results
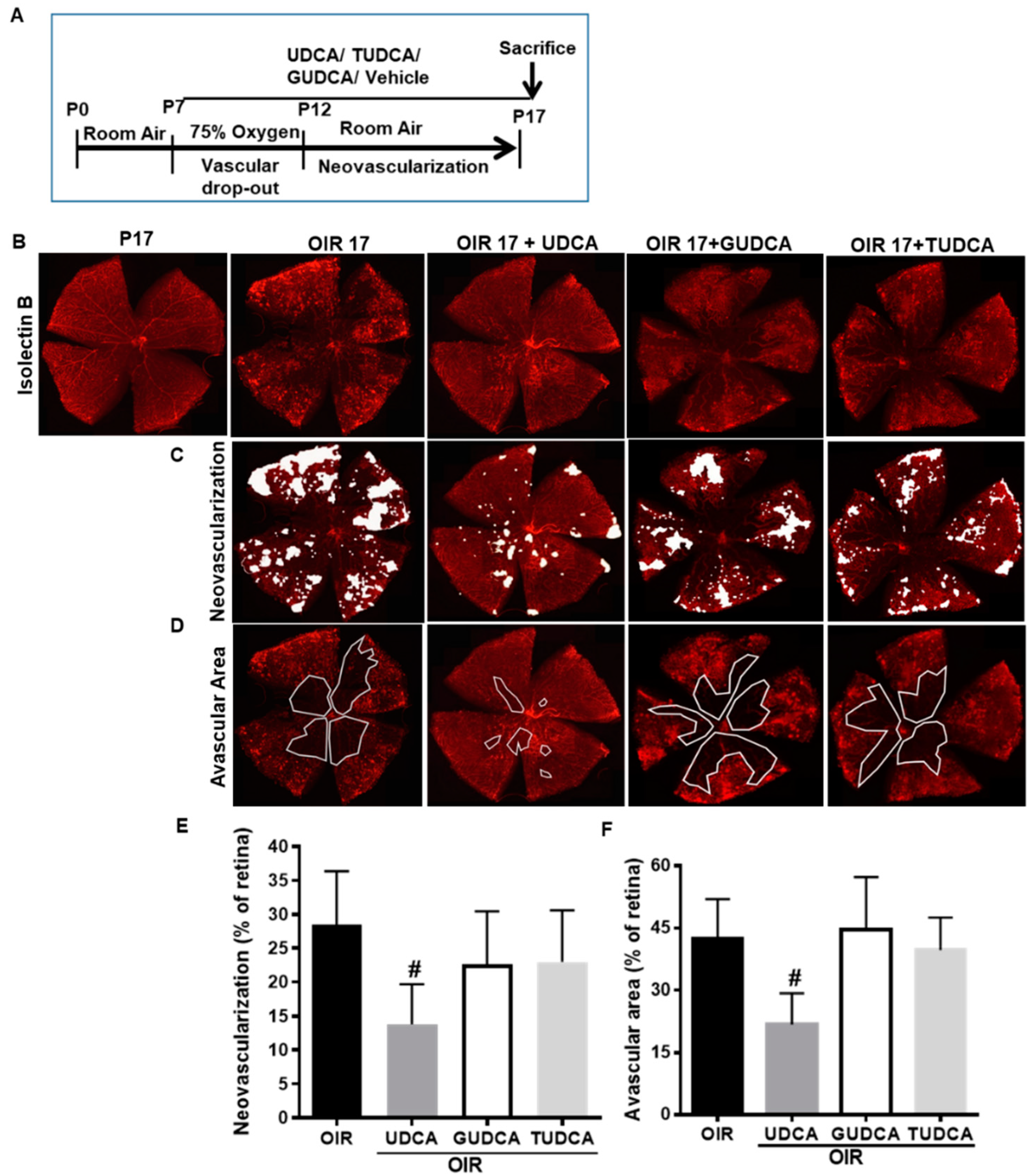
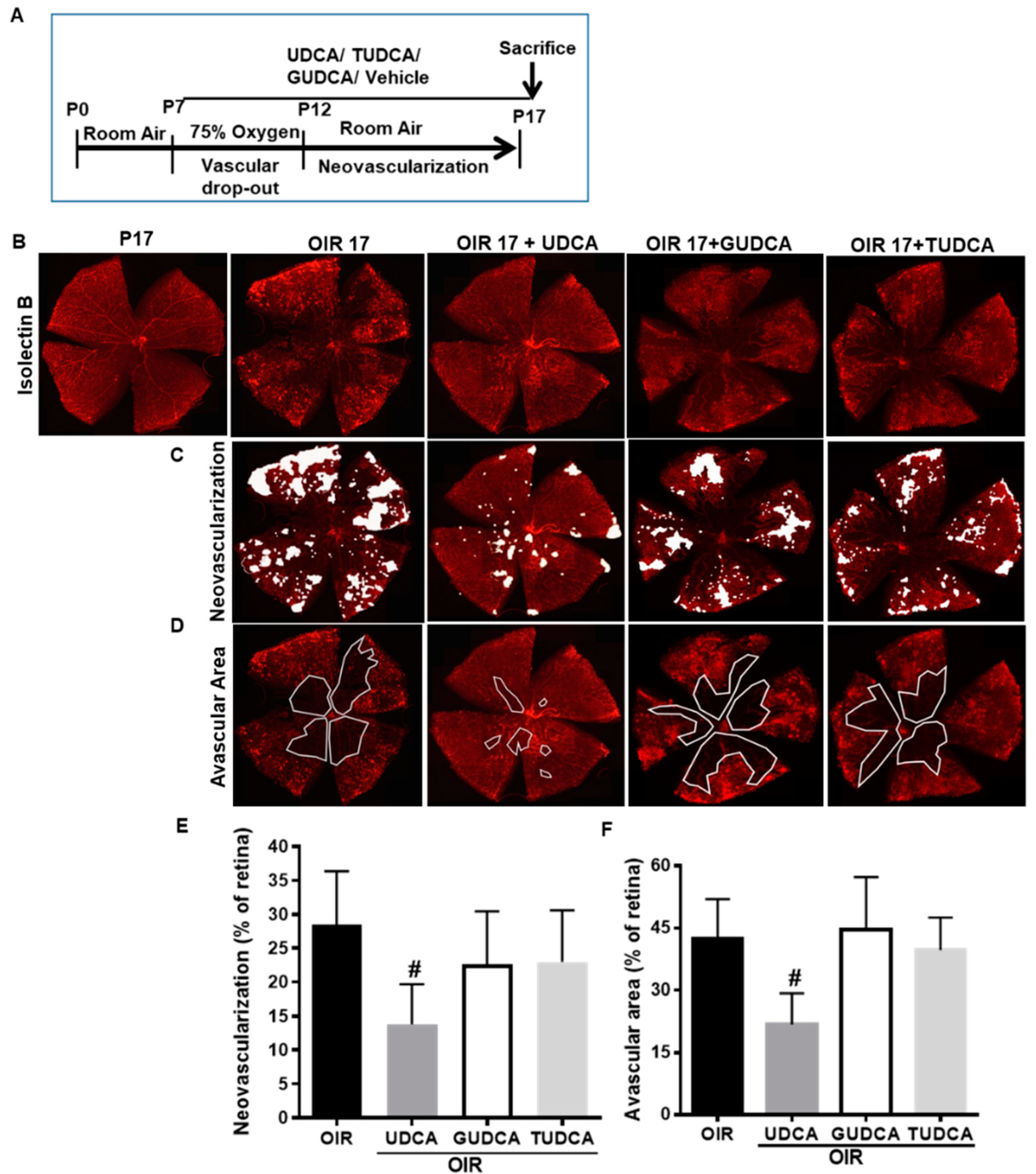
3.1. Differential Effects of Bile Acids on OIR in Mice
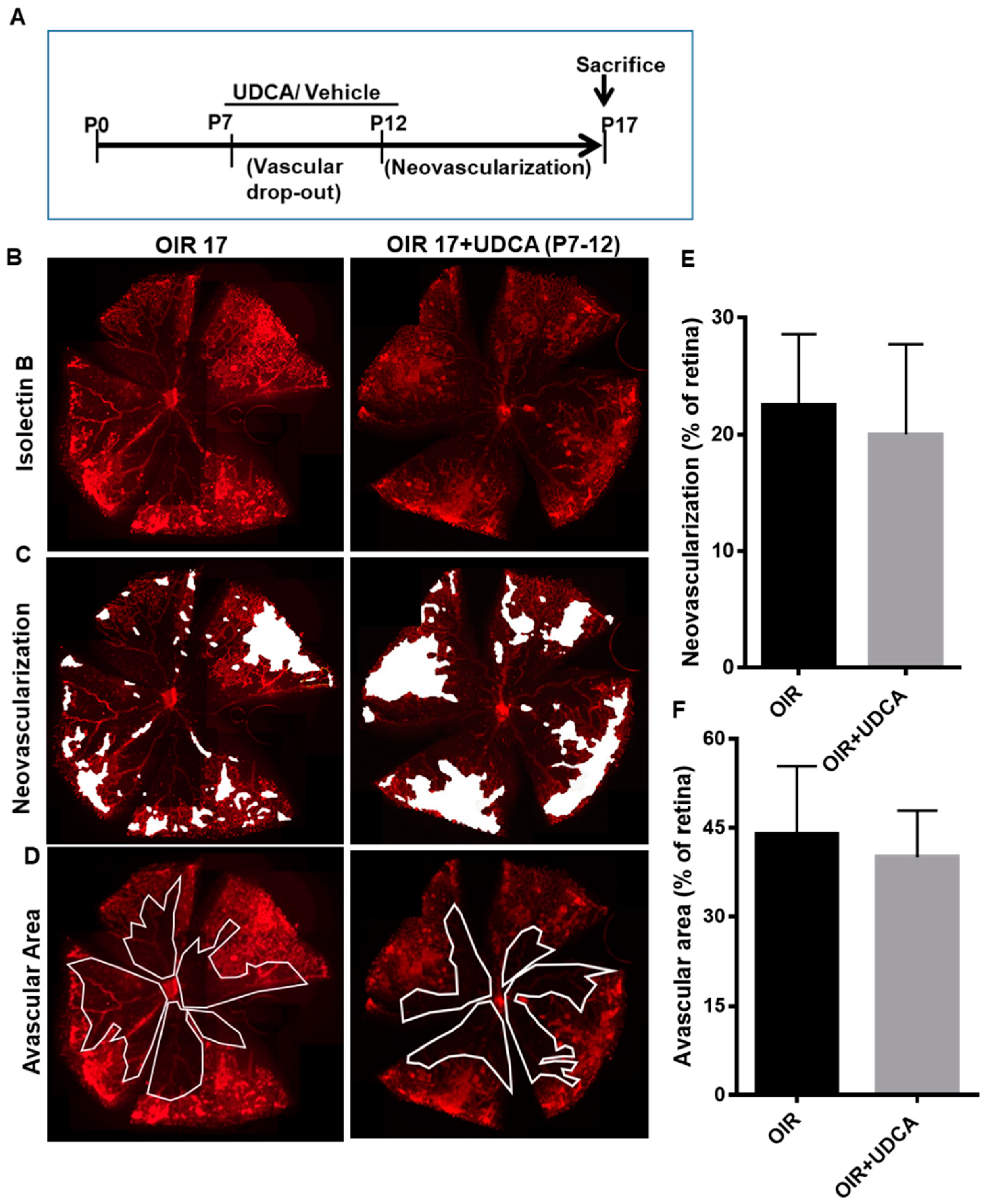
3.2. Effect of UDCA Treatment during Hyperoxic and Hypoxic Phase on OIR in Mice
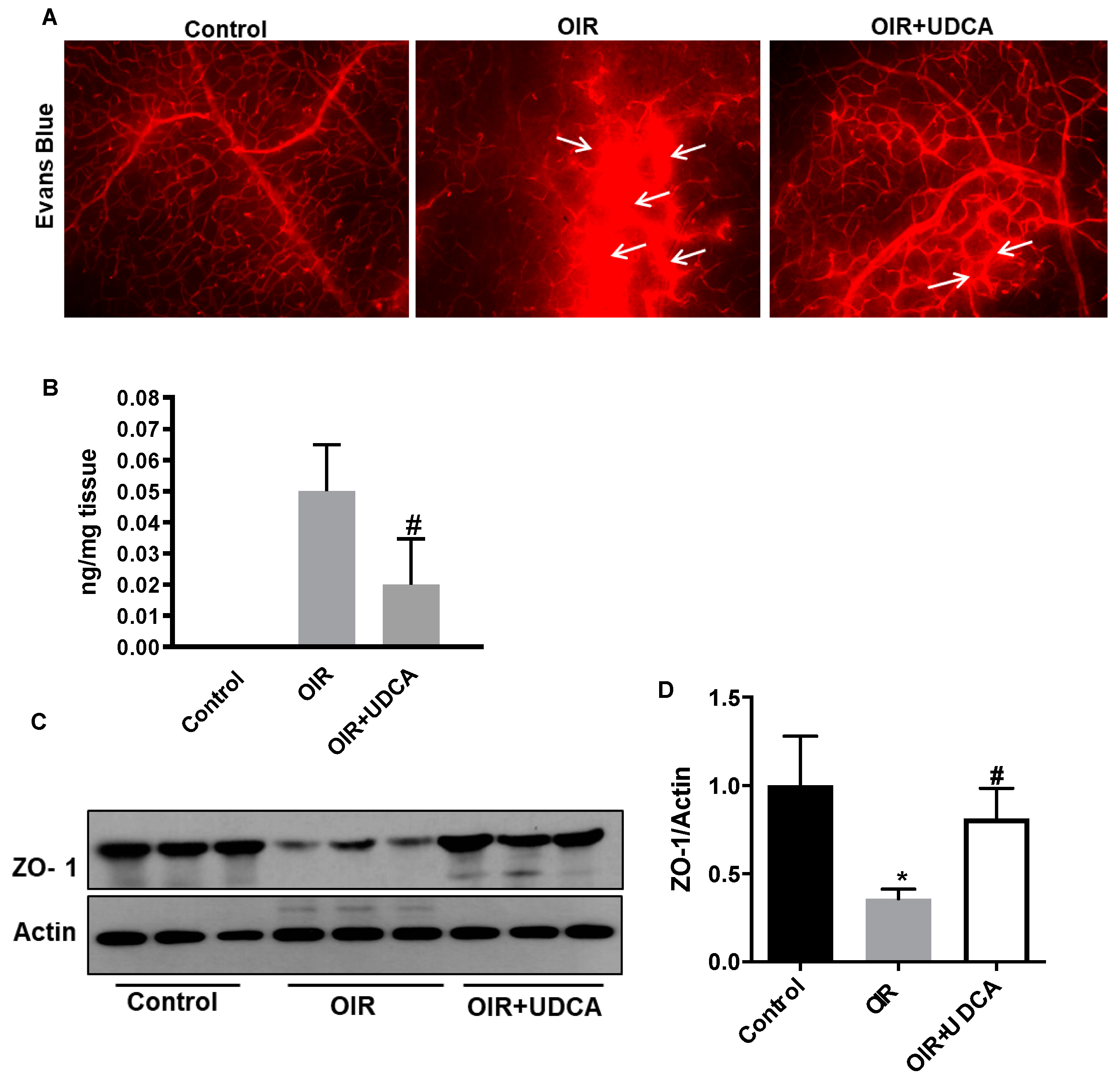
3.3. UDCA Preserves Blood–Retinal Barrier (BRB) in OIR Mice
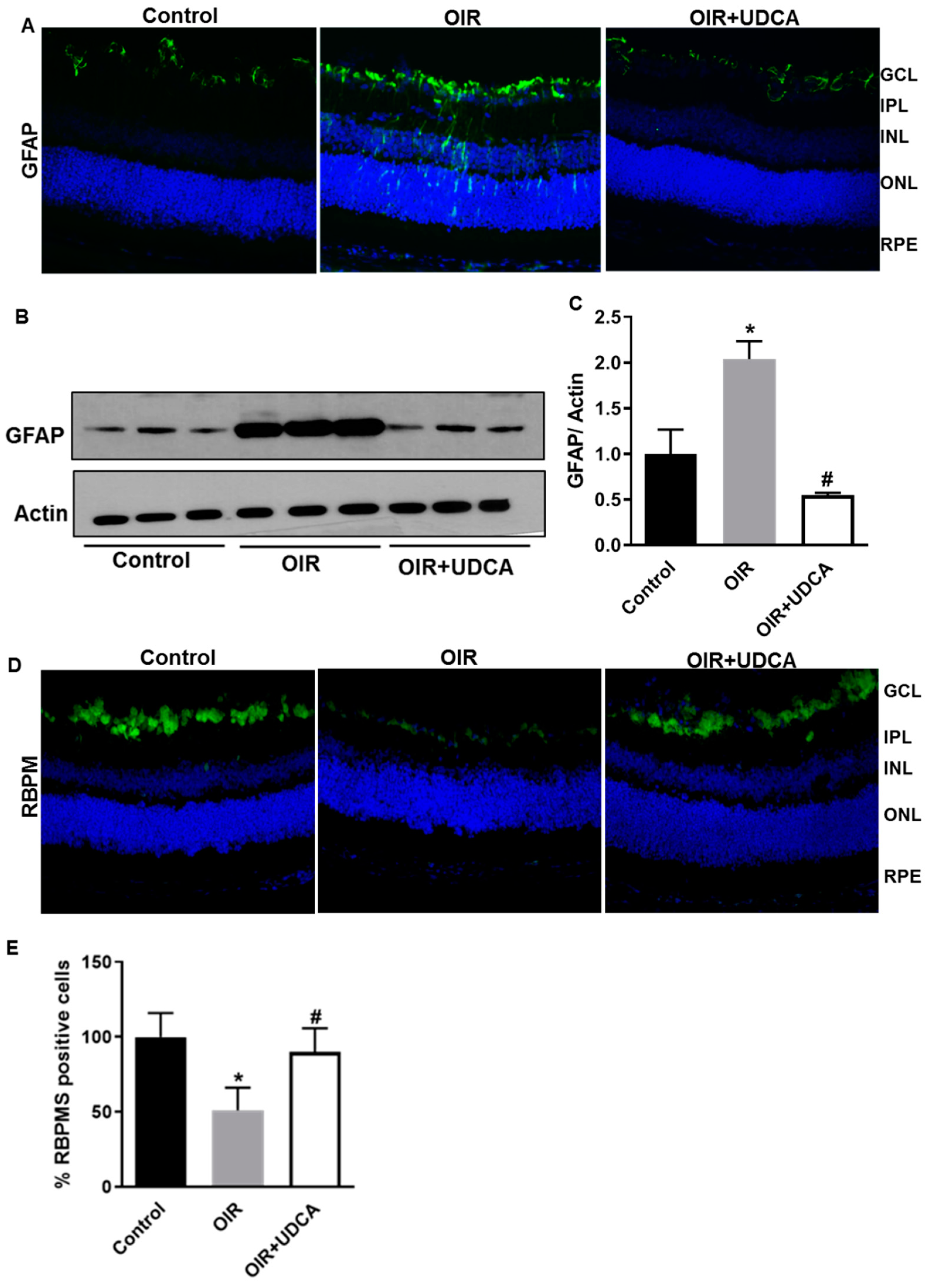
3.4. UDCA Treatment Diminishes Reactive Gliosis and Rescues Neuronal Cells in OIR Mice
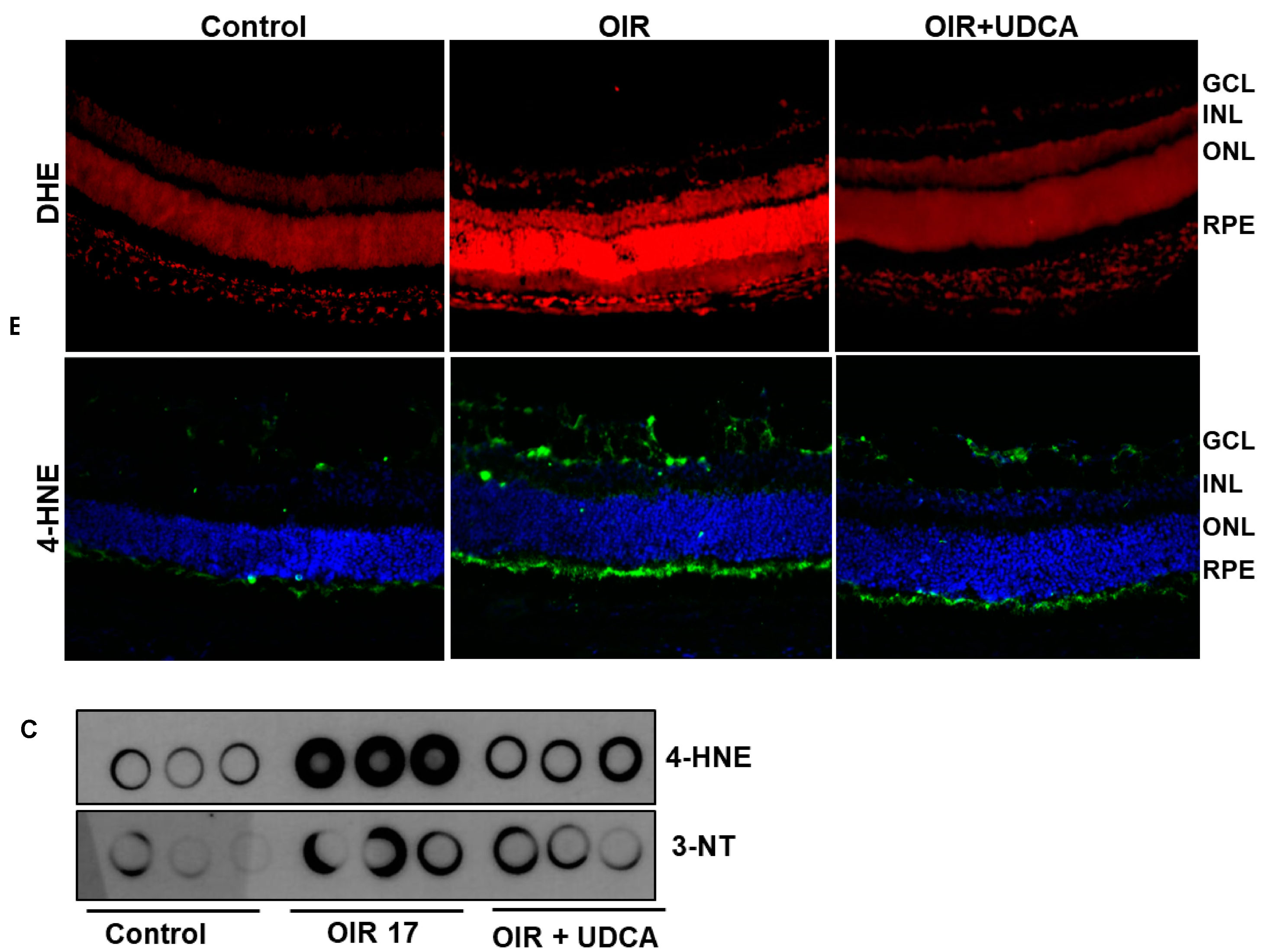
3.5. Effect of UDCA Treatment on Oxidative Stress in OIR Mice
3.6. UDCA Regulates STAT3 Signaling to Reduce Inflammation in OIR Mice
3.7. Effect of UDCA Treatment on VEGF-induced Pro-Angiogenic and Hyperpermeability Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Casteels, I.; Cassiman, C.; Van Calster, J.; Allegaert, K. Educational paper: Retinopathy of prematurity. Eur. J. Pediatr. 2012, 171, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, G.; Filippi, L.; Bagnoli, P.; La Marca, G.; Cristofori, G.; Raffaeli, G.; Padrini, L.; Araimo, G.; Fumagalli, M.; Groppo, M.; et al. The pathophysiology of retinopathy of prematurity: An update of previous and recent knowledge. Acta Ophthalmol. 2013, 92, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.K.; Prabhu, V.; Karandikar, S.S.; Ranjan, R.; Narendran, V.; Kalpana, N. Retinopathy of prematurity: Past, present and future. World J. Clin. Pediatr. 2016, 5, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Park, I.K.; Yu, Y.S. Long term refractive outcome in eyes of preterm infants with and without retinopathy of prematurity: Comparison of keratometric value, axial length, anterior chamber depth, and lens thickness. Br. J. Ophthalmol. 2000, 84, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartnett, M.E.; Gilbert, M.M.; Hirose, T.; Richardson, T.M.; Katsumi, O. Glaucoma as a cause of poor vision in severe retinopathy of prematurity. Graefe’s Arch. Clin. Exp. Ophthalmol. 1993, 231, 433–438. [Google Scholar] [CrossRef]
- Kaiser, R.S.; Trese, M.T.; Williams, G.A.; Cox, M.S., Jr. Adult retinopathy of prematurity: Outcomes of rhegmatogenous retinal detachments and retinal tears. Ophthalmology 2001, 108, 1647–1653. [Google Scholar] [CrossRef]
- Knight-Nanan, D.M.; Algawi, K.; Bowell, R.; O’Keefe, M. Advanced cicatricial retinopathy of prematurity–outcome and complications. Br. J. Ophthalmol. 1996, 80, 343–345. [Google Scholar] [CrossRef]
- Falavarjani, J.K.K.G.; Nguyen, Q.D. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: A review of literature. Eye 2013, 27, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Hartnett, M.E.; Penn, J.S. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 2012, 367, 2515–2526. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, F.M.; Sarici, S.U. Treatment of retinopathy of prematurity: A review of conventional and promising new therapeutic options. Int. J. Ophthalmol. 2013, 6, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Stahl, A.; Krohne, T.U.; Sapieha, P.; Chen, J.; Hellström, A.; Chew, E.; Holz, F.G.; Smith, L.E.H. Lipid metabolites in the pathogenesis and treatment of neovascular eye disease. Br. J. Ophthalmol. 2011, 95, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Bernabe-Garcia, M.; Villegas-Silva, R.; Villavicencio-Torres, A.; Calder, P.C.; Rodriguez-Cruz, M.; Maldonado-Hernández, J.; Macías-Loaiza, D.; López-Alarcón, M.; Inda-Icaza, P.; Cruz-Reynoso, L. Enteral Docosahexaenoic Acid and Retinopathy of Prematurity: A Randomized Clinical Trial. J. Parenter. Enter. Nutr. 2019, 43, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boatright, J.H.; Nickerson, J.M.; Moring, A.G.; Pardue, M.T. Bile acids in treatment of ocular disease. J. Ocul. Boil. Dis. Informatics 2009, 2, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, D.; Arab, J.P.; Arrese, M. UDCA, NorUDCA, and TUDCA in Liver Diseases: A Review of Their Mechanisms of Action and Clinical Applications. Drug Deliv. 2019, 256, 237–264. [Google Scholar] [CrossRef]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Heal. Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.-L.; Zhao, L.; Li, L.; Li, A.-H.; Ye, J.; Yang, L.; Xu, K.-S.; Hou, X.-H. Efficacy and safety of tauroursodeoxycholic acid in the treatment of liver cirrhosis: A double-blind randomized controlled trial. Acta Acad. Med. Wuhan 2013, 33, 189–194. [Google Scholar] [CrossRef]
- Daruich, A.; Picard, E.; Boatright, J.H.; Behar-Cohen, F. Review: The bile acids urso- and tauroursodeoxycholic acid as neuroprotective therapies in retinal disease. Mol. Vis. 2019, 25, 610–624. [Google Scholar]
- Smith, L.E.H.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Bartoli, M.; Al-Shabrawey, M.; Labazi, M.; Behzadian, M.A.; Istanboli, M.; El-Remessy, A.B.; Caldwell, R.W.; Marcus, D.M.; Caldwell, R.B. HMG-CoA reductase inhibitors (statin) prevents retinal neovascularization in a model of oxygen-induced retinopathy. Investig. Opthalmol. Vis. Sci. 2008, 50, 4934–4940. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, M.; Platt, D.H.; Lemtalsi, T.; Gu, X.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. VEGF differentially activates STAT3 in microvascular endothelial cells. FASEB J. 2003, 17, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutsaeva, D.; Thounaojam, M.; Rajpurohit, S.; Powell, F.L.; Martin, P.M.; Goei, S.; Duncan, M.; Bartoli, M. STAT3-mediated activation of miR-21 is involved in down-regulation of TIMP3 and neovascularization in the ischemic retina. Oncotarget 2017, 8, 103568–103580. [Google Scholar] [CrossRef] [PubMed]
- Lamoke, F.; Labazi, M.; Montemari, A.; Parisi, G.; Varano, M.; Bartoli, M. Trans-Chalcone prevents VEGF expression and retinal neovascularization in the ischemic retina. Exp. Eye Res. 2011, 93, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Connor, K.M.; Sapieha, P.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Chen, J.; Guerin, K.I.; Smith, L.E.H. Computer-aided quantification of retinal neovascularization. Angiogenesis 2009, 12, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thounaojam, M.C.; Montemari, A.; Powell, F.L.; Malla, P.; Gutsaeva, D.R.; Bachettoni, A.; Ripandelli, G.; Repossi, A.; Tawfik, A.; Martin, P.M.; et al. Monosodium Urate Contributes to Retinal Inflammation and Progression of Diabetic Retinopathy. Diabetes 2019, 68, 1014–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Xiao, W.; Zhu, X.; Mao, Y.; Liu, X.; Chen, X.; Huang, J.; Tang, S.; Rizzolo, L.J. Differential Expression of Claudins in Retinas during Normal Development and the Angiogenesis of Oxygen-Induced Retinopathy. Investig. Opthalmol. Vis. Sci. 2011, 52, 7556–7564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.-H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Powell, F.L.; Patel, S.; Gutsaeva, D.R.; Tawfik, A.; Smith, S.B.; Nussbaum, J.; Block, N.L.; Martin, P.M.; Schally, A.; et al. Protective effects of agonists of growth hormone-releasing hormone (GHRH) in early experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 13248–13253. [Google Scholar] [CrossRef] [Green Version]
- Promsote, W.; Powell, F.L.; Veean, S.; Thounaojam, M.C.; Markand, S.; Saul, A.; Gutsaeva, D.; Bartoli, M.; Smith, S.B.; Ganapathy, V.; et al. Oral Monomethyl Fumarate Therapy Ameliorates Retinopathy in a Humanized Mouse Model of Sickle Cell Disease. Antioxid. Redox Signal. 2016, 25, 921–935. [Google Scholar] [CrossRef] [Green Version]
- Van Der Wijk, A.-E.; Vogels, I.M.C.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. TNF?-Induced Disruption of the Blood–Retinal Barrier In Vitro Is Regulated by Intracellular 3?,5?-Cyclic Adenosine Monophosphate Levels. Investig. Opthalmol. Vis. Sci. 2017, 58, 3496. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Meng, S.S.; Burnim, S.B.; Smith, L.E.; Lo, A.C.Y. Lutein facilitates physiological revascularization in a mouse model of retinopathy of prematurity. Clin. Exp. Ophthalmol. 2017, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.M.K.; Caprioli, J.; Piri, N. RNA Binding Protein with Multiple Splicing: A New Marker for Retinal Ganglion Cells. Investig. Opthalmol. Vis. Sci. 2010, 51, 1052–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Platt, D.H.; Matragoon, S.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R. Inhibition of NAD(P)H Oxidase Activity Blocks Vascular Endothelial Growth Factor Overexpression and Neovascularization during Ischemic Retinopathy. Am. J. Pathol. 2005, 167, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, M.; Gu, X.; Tsai, N.T.; Venema, R.C.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. Vascular Endothelial Growth Factor Activates STAT Proteins in Aortic Endothelial Cells. J. Boil. Chem. 2000, 275, 33189–33192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, J.K.W.; Liu, J.; Lo, A.C.Y. Vascular and Neuronal Protection in the Developing Retina: Potential Therapeutic Targets for Retinopathy of Prematurity. Int. J. Mol. Sci. 2019, 20, 4321. [Google Scholar] [CrossRef] [Green Version]
- Aranda, J.V.; Qu, J.; Valencia, G.B.; Beharry, K.D. Pharmacologic interventions for the prevention and treatment of retinopathy of prematurity. Semin. Perinatol. 2019, 43, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Feng, J.; Gu, M.-H.; Shi, C.-P.; Zheng, X.-Y.; Zhu, H.-H.; Xie, H.-Y. Analysis on the result of retinopathy of prematurity screening in 1225 premature infants. Zhonghua Er Ke Za Zhi 2010, 48, 829–833. [Google Scholar]
- Dogra, M.R.; Katoch, D.; Dogra, M. An Update on Retinopathy of Prematurity (ROP). Indian J. Pediatr. 2017, 84, 930–936. [Google Scholar] [CrossRef]
- Beli, E.; Yan, Y.; Moldovan, L.; Vieira, C.P.; Gao, R.; Duan, Y.; Prasad, R.; Bhatwadekar, A.; White, F.A.; Townsend, S.D.; et al. Restructuring of the Gut Microbiome by Intermittent Fasting Prevents Retinopathy and Prolongs Survival in db/db Mice. Diabetes 2018, 67, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Mei, X.; Zhang, T.; Lu, B.; Ji, L. Ursodeoxycholic acid ameliorates diabetic retinopathy via reducing retinal inflammation and reversing the breakdown of blood-retinal barrier. Eur. J. Pharmacol. 2018, 840, 20–27. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Elsherbiny, M.; Nussbaum, J.; Othman, A.; Megyerdi, S.; Tawfik, A. Targeting Neovascularization in Ischemic Retinopathy: Recent Advances. Expert Rev. Ophthalmol. 2013, 8, 267–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Li, S.-Y.; Kociok, N.; Wong, D.; Chung, S.K.; Lo, A.C.Y. Aldose Reductase Deficiency Reduced Vascular Changes in Neonatal Mouse Retina in Oxygen-Induced Retinopathy. Investig. Opthalmol. Vis. Sci. 2012, 53, 5698–5712. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Van De Veire, S.; Dalal, M.; Parlier, R.; Semba, R.D.; Carmeliet, P.; Vinores, S.A. Reduced retinal neovascularization, vascular permeability, and apoptosis in ischemic retinopathy in the absence of prolyl hydroxylase-1 due to the prevention of hyperoxia-induced vascular obliteration. Investig. Opthalmol. Vis. Sci. 2011, 52, 7565–7573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, D.; Bartoli, M.; Elremessy, A.; Al-Shabrawey, M.; Lemtalsi, T.; Fulton, D.; Caldwell, R. Peroxynitrite increases VEGF expression in vascular endothelial cells via STAT3. Free. Radic. Boil. Med. 2005, 39, 1353–1361. [Google Scholar] [CrossRef]
- Penn, J.S.; Madan, A.; Caldwell, R.; Bartoli, M.; Hartnett, M.; Caldwell, R. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharjan, P.; Kim, D.; Jin, M.; Ko, H.J.; Song, Y.H.; Lee, Y.; Ahn, B.-N.; Shin, M.C.; Min, K.A.; Yang, J.; et al. Preclinical Evaluation of UDCA-Containing Oral Formulation in Mice for the Treatment of Wet Age-Related Macular Degeneration. Pharmaceutics 2019, 11, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinot, E.; Sèdes, L.; Baptissart, M.; Lobaccaro, J.-M.; Caira, F.; Beaudoin, C.; Volle, D.H. Bile acids and their receptors. Mol. Asp. Med. 2017, 56, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Nishimaki-Mogami, T.; Yamaguchi, M.; Teraoka, F.; Kaneko, T.; Une, M. Effects of chemical modification of ursodeoxycholic acid on TGR5 activation. Boil. Pharm. Bull. 2011, 34, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Carino, A.; Biagioli, M.; Marchianò, S.; Fiorucci, C.; Zampella, A.; Monti, M.C.; Scarpelli, P.; Ricci, P.; Distrutti, E.; Fiorucci, S. Ursodeoxycholic acid is a GPBAR1 agonist and resets liver/intestinal FXR signaling in a model of diet-induced dysbiosis and NASH. Biochim. Biophys. Acta Mol. Cell Boil. Lipids 2019, 1864, 1422–1437. [Google Scholar] [CrossRef]
- Campana, G.; Pasini, P.; Roda, A.; Spampinato, S. Regulation of ileal bile acid-binding protein expression in Caco-2 cells by ursodeoxycholic acid: Role of the farnesoid X receptor. Biochem. Pharmacol. 2005, 69, 1755–1763. [Google Scholar] [CrossRef]
- Yanguas-Casas, N.; Barreda-Manso, M.A.; Nieto-Sampedro, M.; Romero-Ramírez, L. TUDCA: An Agonist of the Bile Acid Receptor GPBAR1/TGR5 With Anti-Inflammatory Effects in Microglial Cells. J. Cell Physiol. 2017, 232, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.A.; Rivera, A.; Cai, J.; Patterson, A.D.; Theriot, C.M. Secondary bile acid ursodeoxycholic acid (UDCA) alters weight, the gut microbiota, and the bile acid pool in conventional mice. bioRxiv 2019, 698795. [Google Scholar] [CrossRef]
- Kang, H.G.; Choi, E.Y.; Byeon, S.H.; Kim, S.S.; Koh, H.-J.; Lee, S.C.; Kim, M. Anti-vascular Endothelial Growth Factor Treatment of Retinopathy of Prematurity: Efficacy, Safety, and Anatomical Outcomes. Korean J. Ophthalmol. 2018, 32, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thounaojam, M.C.; Jadeja, R.N.; Rajpurohit, S.; Gutsaeva, D.R.; Stansfield, B.K.; Martin, P.M.; Bartoli, M. Ursodeoxycholic Acid Halts Pathological Neovascularization in a Mouse Model of Oxygen-Induced Retinopathy. J. Clin. Med. 2020, 9, 1921. https://doi.org/10.3390/jcm9061921
Thounaojam MC, Jadeja RN, Rajpurohit S, Gutsaeva DR, Stansfield BK, Martin PM, Bartoli M. Ursodeoxycholic Acid Halts Pathological Neovascularization in a Mouse Model of Oxygen-Induced Retinopathy. Journal of Clinical Medicine. 2020; 9(6):1921. https://doi.org/10.3390/jcm9061921
Chicago/Turabian StyleThounaojam, Menaka C., Ravirajsinh N. Jadeja, Shubhra Rajpurohit, Diana R. Gutsaeva, Brian K. Stansfield, Pamela M. Martin, and Manuela Bartoli. 2020. "Ursodeoxycholic Acid Halts Pathological Neovascularization in a Mouse Model of Oxygen-Induced Retinopathy" Journal of Clinical Medicine 9, no. 6: 1921. https://doi.org/10.3390/jcm9061921
APA StyleThounaojam, M. C., Jadeja, R. N., Rajpurohit, S., Gutsaeva, D. R., Stansfield, B. K., Martin, P. M., & Bartoli, M. (2020). Ursodeoxycholic Acid Halts Pathological Neovascularization in a Mouse Model of Oxygen-Induced Retinopathy. Journal of Clinical Medicine, 9(6), 1921. https://doi.org/10.3390/jcm9061921