Hepatitis C Virus Influences HIV-1 Viral Splicing in Coinfected Patients

 , ,
, ,  ,
on behalf of COVIHEP
,
on behalf of COVIHEP
Abstract
1. Introduction
2. Methods
2.1. Patients
2.2. Ethical Statement
2.3. Isolation of PBMCs and Total RNA Extraction
2.4. Quantification of HIV-1 Proviral DNA Integration
2.5. HIV Viral Splicing Analysis by qPCR
2.6. Quantification of Tat mRNA Expression
2.7. Statistical Analysis
3. Results
3.1. Study Patients
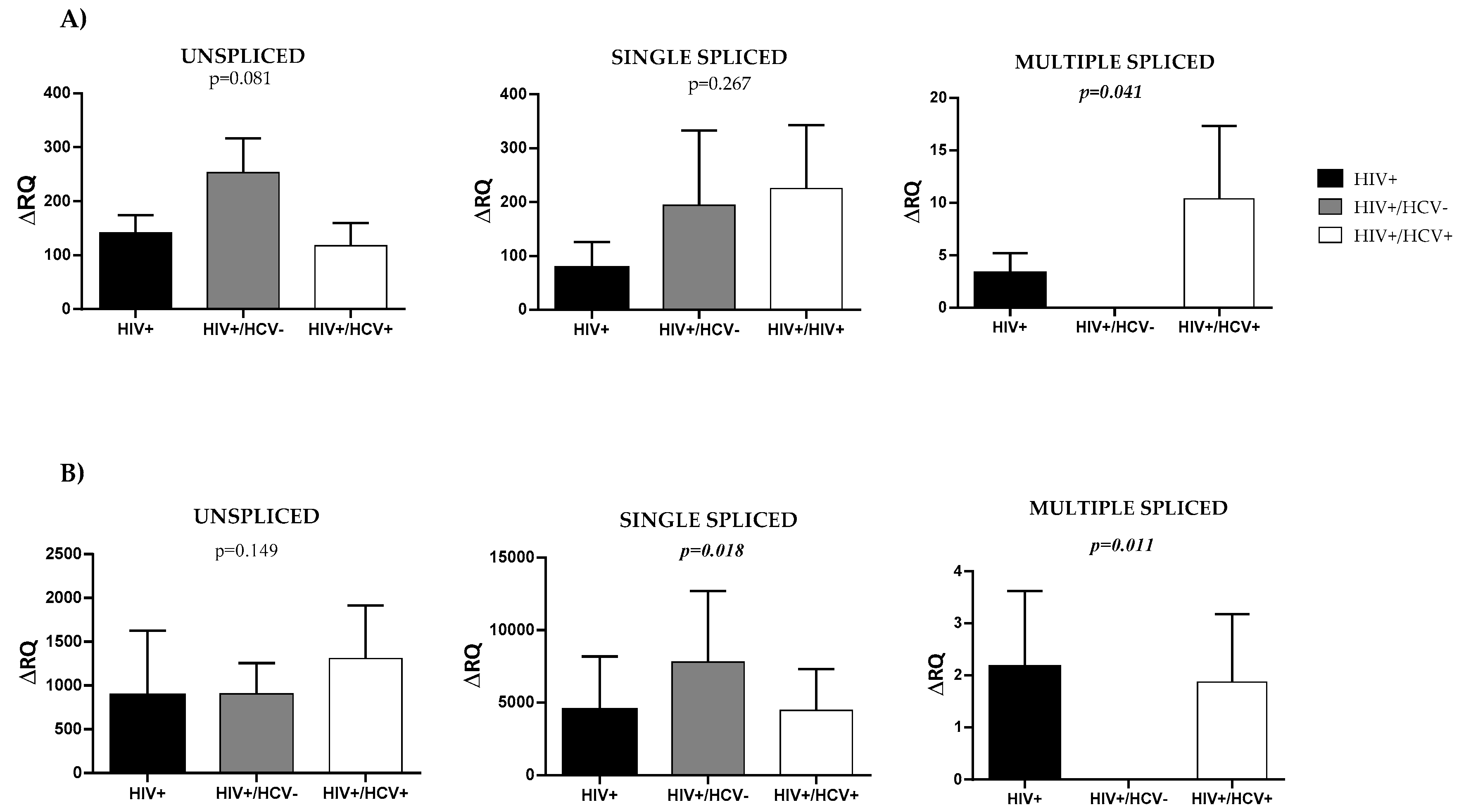
3.2. HIV Splicing Analysis
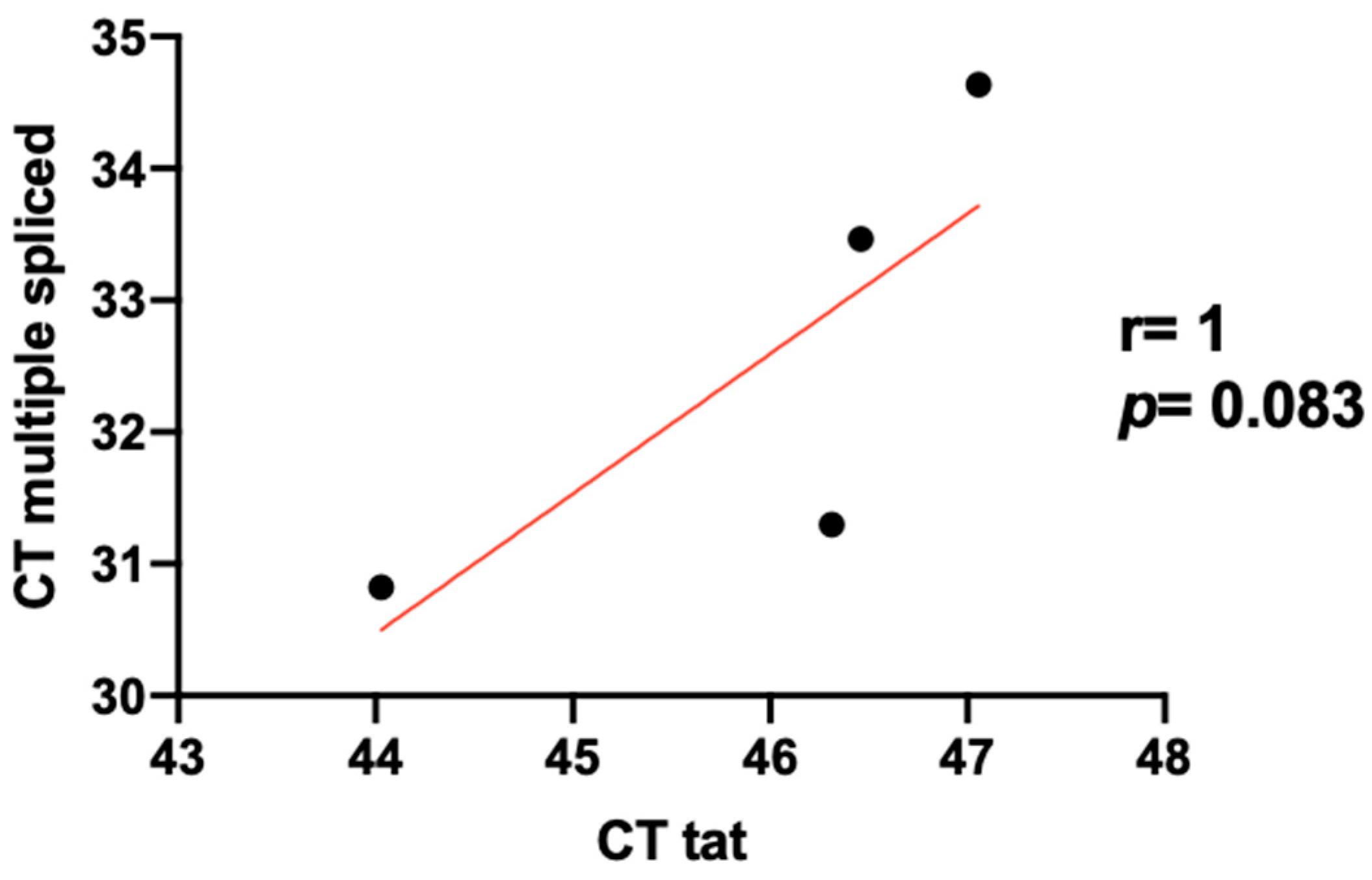
3.3. Level Expression of Tat in HIV/HCV Coinfection
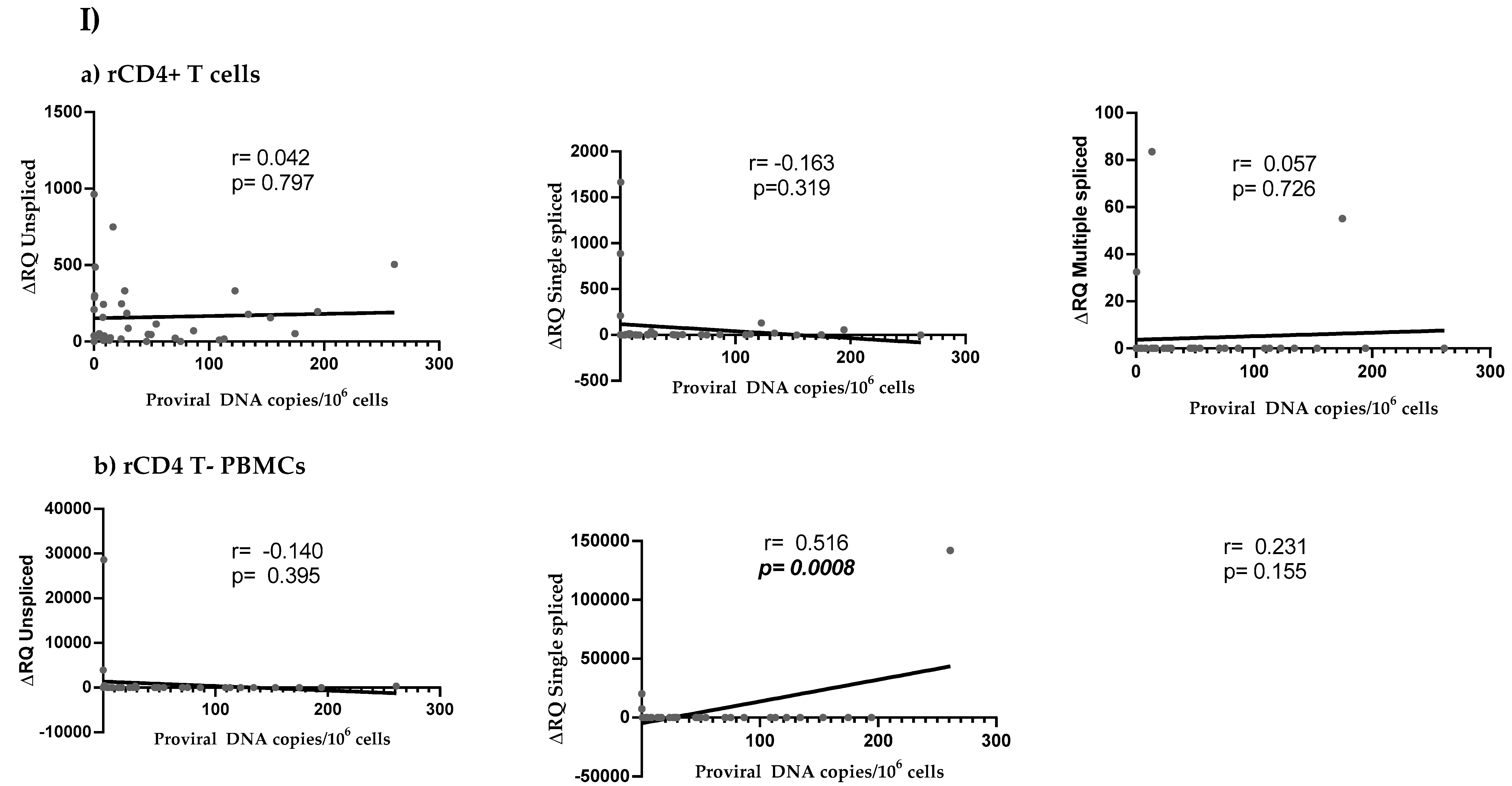
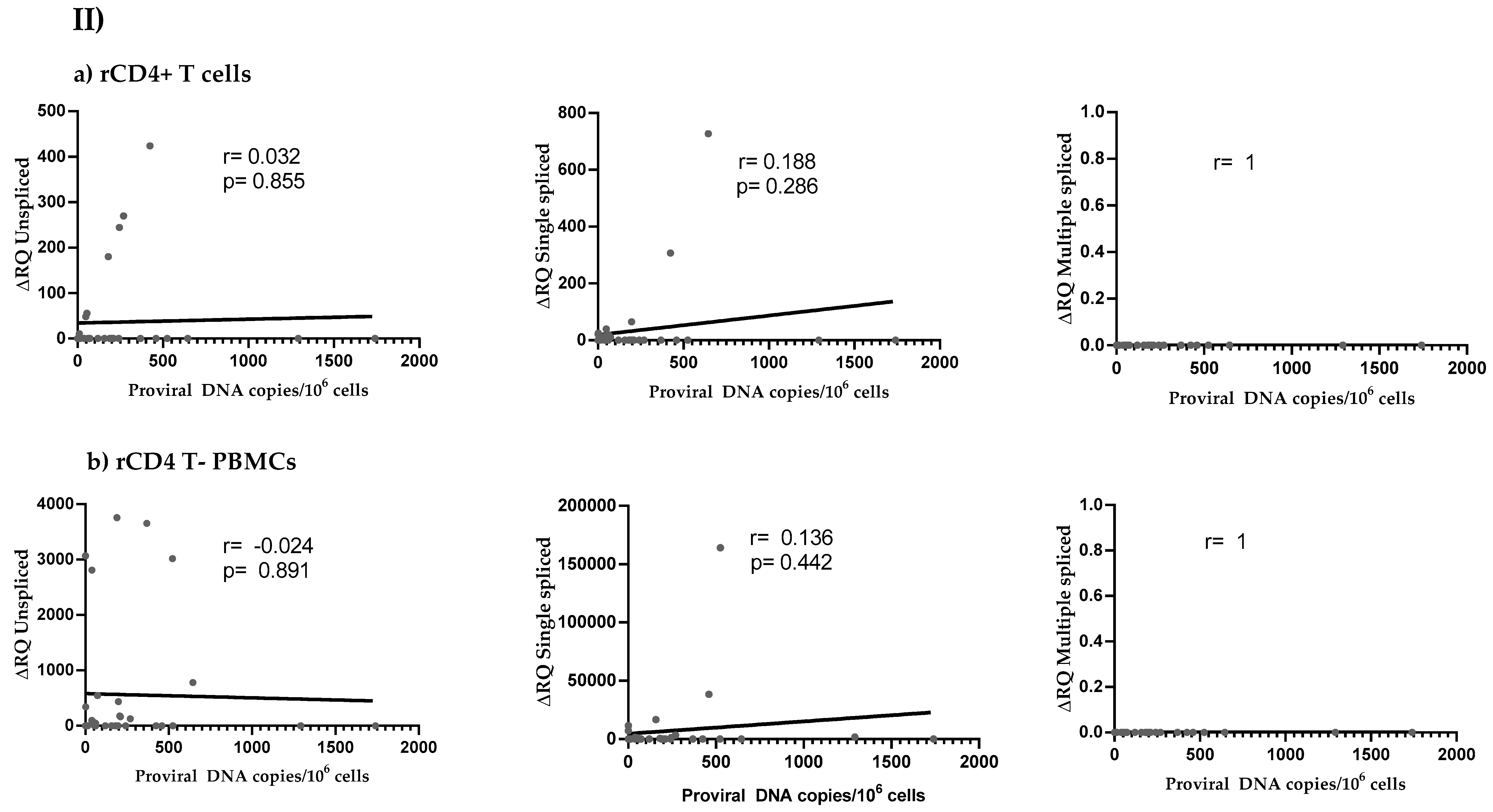
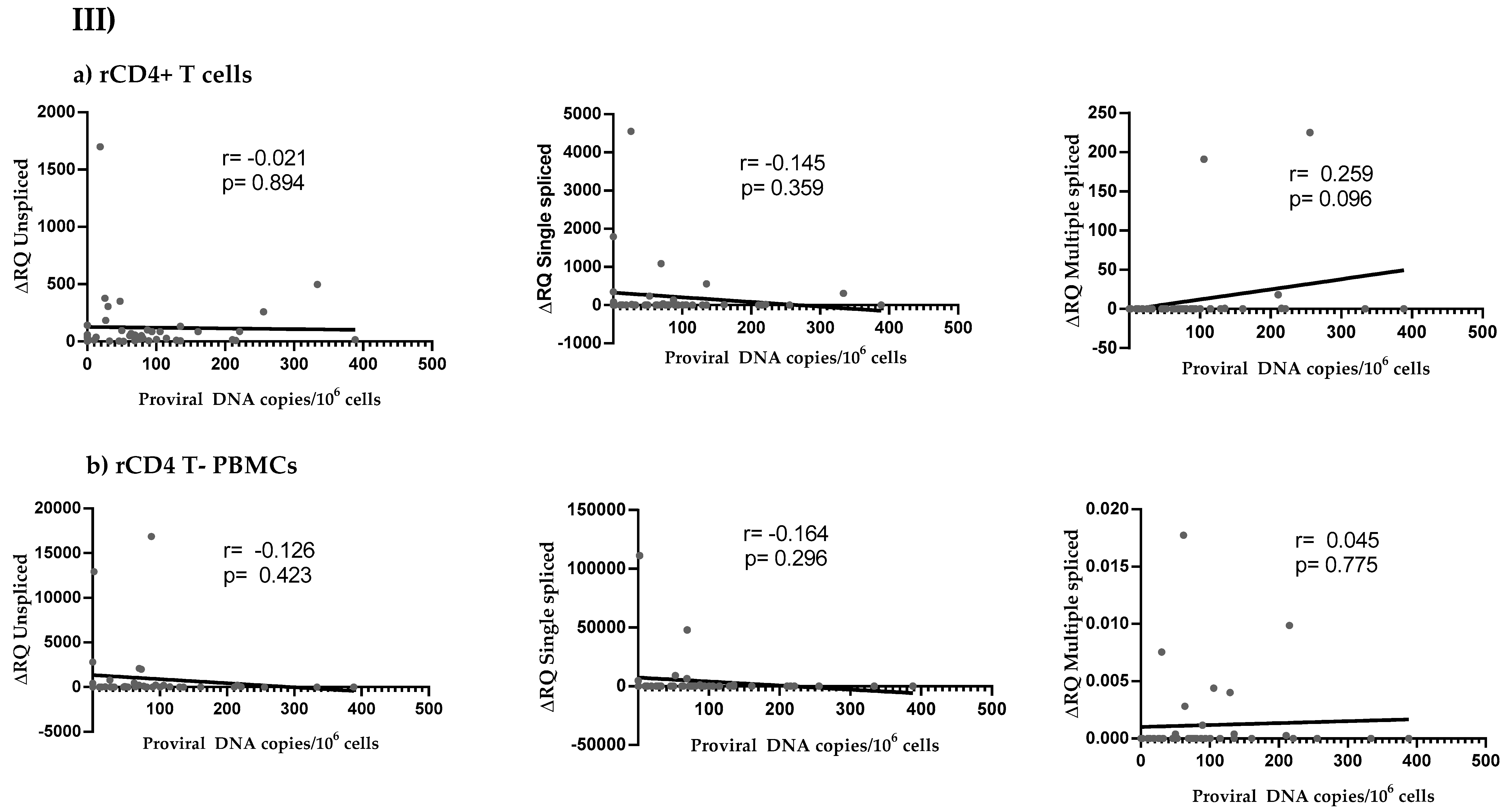
3.4. HIV Reservoir Size and HIV Splicing Correlation
4. Discussion
Author Contributions
Funding
Acknowledgments
- Servicio de Medicina Interna, Hospital Puerta de Hierro, 28222 Madrid, Spain.
- AIDS Immunopathology Unit, National Center of Microbiology, Institute of Health Carlos III 28220 Majadahonda, Madrid, Spain.
- Unidad VIH, Servicio de Medicina Interna, Instituto de Investigación Biomédica del Hospital Doce de Octubre (imas12), 28041 Madrid, Spain.
- Laboratory of Reference and Research on Viral Hepatitis, National Center of Microbiology, Institute of Health Carlos III, 28220 Majadahonda, Madrid, Spain.
- Instituto de Investigación Sanitaria Hospital de la Paz (IdiPAZ), 28046 Madrid, Spain.
- Department of Infectious Diseases, Infanta Leonor Hospital, 28031 Madrid, Spain.
- Servicio de Medicina Interna-Infecciosas, Hospital Universitario de La Princesa, 28006 Madrid, Spain.
- The Roslin Institute, Edinburgh, UK.
- National Institute of Agricultural and Food Research and Technology, Madrid, Spain.
- Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Conflicts of Interest
Appendix A
- -
- Hospital Universitario 12 de octubre (Madrid-Spain): Laura Bermejo-Plaza; Otilia Bisbal; Lourdes Domínguez-Domínguez; María Lagarde; Mariano Matarranz; Federico Pulido; Rafa Rubio; Mireia Santacreu.
- -
- Hospital Universitario Infanta Leonor (Madrid-Spain): Guillermo Cuevas; Victorino Diez-Viñas; Pablo Ryan; Jesús Troya.
- -
- Hospital Universitario La Paz (Madrid-Spain): Juan Miguel Castro-Álvarez; Marta Gálvez-Charro; Luz Martín-Carbonero; Mario Mayoral-Muñoz.
- -
- Hospital Universitario La Princesa (Madrid-Spain): Ignacio de los Santos; Lucio García-Fraile; Jesús Sanz-Sanz.
- -
- Hospital Universitario Puerta del Hierro Majadahonda (Madrid-Spain): Alfonso Ángel-Moreno; Sara de la Fuente Moral.
- -
- Instituto de salud Carlos III Majadahonda (Madrid-Spain): José Alcamí; Sonia Arca-Lafuente; Verónica Briz, Oscar Brochado; Celia Crespo-Bermejo; Mayte Coiras; Alicia Gómez-Sanz, Amanda Fernández-Rodríguez; Irene Mate-Cano; Paula Martínez-Román; Elena Mateo; Salvado Resino; Marta Sánchez-Carrillo; Daniel Valle-Miralles.
- -
- Research Institute for Medicines (Lisboa-Portugal): Claudia Palladino; Nuno Taveira
- -
- The Roslin Institute (Edinburgh-UK) and CENIDCI, Centro Nacional de Investigación y Desarrollo del Cerdo Ibérico of INIA (Madrid-Spain): María Muñoz-Muñoz.
Appendix B
| PRIMER NAME | 5’-3’ PRIMER SEQUENCE | ORIENTATION | PURPOSE |
| Tat FW | ATGGAGCCAGTAGATCCTA | fwd | Tat |
| Tat 72qpcr RV | AGACAGCGACGAAGACCTC | rev | Tat |
| b-act s | AGGCCCAGAGCAAGAGAGGCA | fwd | β-actin (GeneID: 60) |
| b-act as | CGCAGCTCATTGTAGAAGGTGTGGT | fwd | β-actin (GeneID: 60) |
References
- UNAIDS Data 2018 [Internet]. Unaids.org. 2019. Available online: https://www.unaids.org/en (accessed on 26 February 2019).
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Coiras, M.; López-Huertas, M.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- Global Hepatitis Report, 2017 [Internet]. World Health Organization. 2019. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 26 February 2019).
- Platt, L.; Easterbrook, P.; Gower, E.; McDonald, B.; Sabin, K.; McGowan, C.; Yanny, I.; Razavi, H.; Vickerman, P. Prevalence and burden of HCV co-infection in people living with HIV: A global systematic review and meta-analysis. Lancet Infect. Dis. 2016, 16, 797–808. [Google Scholar] [CrossRef]
- Graham, C.S.; Baden, L.R.; Yu, E.; Mrus, J.M.; Carnie, J.; Heeren, T.; Koziel, M.J. Influence of Human Immunodeficiency Virus Infection on the Course of Hepatitis C Virus Infection: A Meta-Analysis. Clin. Infect. Dis. 2001, 33, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Rallón, N.; García, M.; García-Samaniego, J.; Rodríguez, N.; Cabello, A.; Restrepo, C.; Álvarez, B.; García, R.; Górgolas, M.; Benito, J.M. HCV coinfection contributes to HIV pathogenesis by increasing immune exhaustion in CD8 T-cells. PLoS ONE 2017, 12, e0173943. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, D.; Astemborski, J.; Rai, R.; Anania, F.; Schaeffer, M.; Galai, N.; Galai, N.; Nolt, K.; Nelson, K.E.; Strathdee, S.A.; et al. The Natural History of Hepatitis C Virus Infection. JAMA 2000, 284, 450–456. [Google Scholar] [CrossRef] [PubMed]
- López-Huertas, M.R.; Palladino, C.; Garrido-Arquero, M.; Esteban-Cartelle, B.; Sánchez-Carrillo, M.; Martínez-Román, P.; Martín-Carbonero, L.; Ryan, P.; Domínguez-Domínguez, L.; De los Santos, I.; et al. HCV-coinfection is related to an increased HIV-1 reservoir size in cART-treated HIV patients: A cross-sectional study. Sci. Rep. 2019, 9, 5606. [Google Scholar] [CrossRef]
- Schwartz, S.; Felber, B.K.; Benko, D.M.; Fenyö, E.M.; Pavlakis, G.N. Cloning and functional analysis of multiply spliced mRNA species of human immunodeficiency virus type 1. J. Virol. 1990, 64, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Purcell, D.F.; Martin, M.A. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef]
- Carrera, C.; Pinilla, M.; Pérez-Alvarez, L.; Thomson, M.M. Identification of unusual and novel HIV type 1 spliced transcripts generated in vivo. AIDS Res. Hum. Retrovir. 2010, 26, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Epstein, F.; Epstein, F.; Greene, W. The Molecular Biology of Human Immunodeficiency Virus Type 1 Infection. N. Eng. J. Med. 1991, 324, 308–317. [Google Scholar] [CrossRef]
- Jablonski, J.; Amelio, A.; Giacca, M.; Caputi, M. The transcriptional transactivator Tat selectively regulates viral splicing. Nucleic Acids Res. 2009, 38, 1249–1260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mueller, N.; Pasternak, A.; Klaver, B.; Cornelissen, M.; Berkhout, B.; Das, A. The HIV-1 Tat Protein Enhances Splicing at the Major Splice Donor Site. J. Virol. 2018, 92, e01855-17. [Google Scholar] [CrossRef]
- Tazi, J.; Bakkour, N.; Marchand, V.; Ayadi, L.; Aboufirassi, A.; Branlant, C. Alternative splicing: Regulation of HIV-1 multiplication as a target for therapeutic action. FEBS J. 2010, 277, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Karn, J.; Stoltzfus, C. Transcriptional and Posttranscriptional Regulation of HIV-1 Gene Expression. CSH Perspect. Med. 2011, 2, a006916. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, Z.; Hull, R. Can the HIV-1 splicing machinery be targeted for drug discovery? HIV/AIDS Res. Palliat. Care 2017, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- McCracken, S.; Fong, N.; Rosonina, E.; Yankulov, K.; Brothers, G.; Siderovski, D.; Hessel, A.; Foster, S.; Shuman, S.; Bentley, D.L. 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997, 11, 3306–3318. [Google Scholar] [CrossRef]
- De la Mata, M.; Muñoz, M.J.; Alló, M.; Fededa, J.P.; Schor, I.E.; Kornblihtt, A.R. RNA Polymerase II Elongation at the Crossroads of Transcription and Alternative Splicing. Genet. Res. Int. 2011, 2011, 309865. [Google Scholar] [CrossRef]
- Berkhout, B.; Jeang, K.T. Trans activation of human immunodeficiency virus type 1 is sequence specific for both the single-stranded bulge and loop of the trans-acting-responsive hairpin: A quantitative analysis. J. Virol. 1989, 63, 5501–5504. [Google Scholar] [CrossRef]
- Kao, S.; Calman, A.; Luciw, P.; Peterlin, B. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Rice, A. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Design. 2017, 23, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Oh, H.; Wanless, I.; Lee, S.; Han, K.; Park, Y. The Laennec staging system for histological sub-classification of cirrhosis is useful for stratification of prognosis in patients with liver cirrhosis. J. Hepatol. 2012, 57, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Field, N.; Cohen, T.; Struelens, M.J.; Palm, D.; Cookson, B.; Glynn, J.R.; Gallo, V.; Ramsay, M.; Sonnenberg, P.; MacCannell, D.; et al. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): An extension of the STROBE statement. Lancet Infect. Dis. 2014, 14, 341–352. [Google Scholar] [CrossRef]
- Mohammadi, P.; Desfarges, S.; Bartha, I.; Joos, B.; Zangger, N.; Munoz, M.; Günthard, H.F.; Beerenwinkel, N.; Telenti, A.; Ciuffi, A. 24 Hours in the Life of HIV-1 in a T Cell Line. PLoS Pathog. 2013, 9, e1003161. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Anderson, C.M.; Vargas, M.V.; Richman, D.D.; Little, S.J.; Morris, S.R.; Smith, D.M. Cytomegalovirus DNA in Semen and Blood Is Associated with Higher Levels of Proviral HIV DNA. J. Infect. Dis. 2012, 207, 898–902. [Google Scholar] [CrossRef]
- Manns, M.; Buti, M.; Gane, E.; Pawlotsky, J.; Razavi, H.; Terrault, N. Hepatitis C virus infection. Nat. Rev. Dis Primers. 2017, 3. [Google Scholar] [CrossRef]
- Pasternak, A.; Berkhout, B. What do we measure when we measure cell-associated HIV RNA. Retrovirology. 2018, 15. [Google Scholar] [CrossRef]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.; Razavi, H.; Terrault, N.; Younossi, Z. A Novel Assay to Measure the Magnitude of the Inducible Viral Reservoir in HIV-infected Individuals. EBioMedicine 2015, 2, 874–883. [Google Scholar]
- Zanchetta, M.; Walker, S.; Burighel, N.; Bellanova, D.; Rampon, O.; Giaquinto, C.; Rossi, A.D. Long-Term Decay of the HIV-1 Reservoir in HIV-1–Infected Children Treated with Highly Active Antiretroviral Therapy. J. Infect. Dis. 2006, 193, 1718–1727. [Google Scholar] [CrossRef]
- Schmid, A.; Gianella, S.; von Wyl, V.; Metzner, K.J.; Scherrer, A.U.; Niederöst, B.; Althaus, C.F.; Rieder, P.; Grube, C.; Joos, B.; et al. Profound Depletion of HIV-1 Transcription in Patients Initiating Antiretroviral Therapy during Acute Infection. PLoS ONE 2010, 5, e13310. [Google Scholar] [CrossRef]
- Vesanen, M.; Markowitz, M.; Cao, Y.; Ho, D.; Saksela, K. Human Immunodeficiency Virus Type-1 mRNA Splicing Pattern in Infected Persons Is Determined by the Proportion of Newly Infected Cells. Virology 1997, 236, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Uprety, P.; Chadwick, E.G.; Rainwater-Lovett, K.; Ziemniak, C.; Luzuriaga, K.; Capparelli, E.V.; Yenokyan, G.; Persaud, D. Cell-Associated HIV-1 DNA and RNA Decay Dynamics During Early Combination Antiretroviral Therapy in HIV-1-Infected Infants. Clin. Infect. Dis. 2015, 61, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Joos, B.; Niederost, B.; Weber, R.; Gunthard, H.; Fischer, M. Productive Human Immunodeficiency Virus Type 1 Infection in Peripheral Blood Predominantly Takes Place in CD4/CD8 Double-Negative T Lymphocytes. J. Virol. 2007, 81, 9693–9706. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Joos, B.; Niederöst, B.; Kaiser, P.; Hafner, R.; von Wyl, V.; Ackermann, M.; Weber, R.; Günthard, H.F. Biphasic decay kinetics suggest progressive slowing in turnover of latently HIV-1 infected cells during antiretroviral therapy. Retrovirology 2008, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Joshi, S.K.; Kim, N.; Li, P.; Lampiris, H.; Liu, H.; Rice, A.; Wong, J.K. Investigating the mechanisms that control HIV transcription and latency in vivo. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 22–25 February 2016. [Google Scholar]
- Lewin, S.; Rouzioux, C. HIV cure and eradication: How will we get from the laboratory to effective clinical trials? AIDS 2011, 25, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Gatignol AJeang, K.T. Tat as a transcriptional activator and a potential therapeutic target for HIV-I. Adv. Pharmacol. 2000, 48, 209–227. [Google Scholar]
- Lewin, S.R.; Vesanen, M.; Kostrikis, L.; Hurley, A.; Duran, M.; Zhang, L.; Ho, D.D.; Markowitz, M. Use of Real-Time PCR and Molecular Beacons To Detect Virus Replication in Human Immunodeficiency Virus Type 1-Infected Individuals on Prolonged Effective Antiretroviral Therapy. J. Virol. 1999, 73, 6099–6103. [Google Scholar] [CrossRef]
- Pasternak, A.; Jurriaans, S.; Bakker, M.; Prins, J.; Berkhout, B.; Lukashov, V. Cellular Levels of HIV Unspliced RNA from Patients on Combination Antiretroviral Therapy with Undetectable Plasma Viremia Predict the Therapy Outcome. PLoS ONE 2009, 4, e8490. [Google Scholar] [CrossRef]
- Diepolder, H. New insights into the immunopathogenesis of chronic hepatitis C. Antivir. Res. 2009, 82, 103–109. [Google Scholar] [CrossRef]
- Garten, R.; Lai, S.; Zhang, J.; Liu, W.; Chen, J.; Yu, X. Factors influencing a low rate of hepatitis C viral RNA clearance in heroin users from Southern China. World J. Gastroenterol. 2008, 14, 1878. [Google Scholar] [CrossRef]
- Grebely, J.; Raffa, J.; Lai, C.; Krajden, M.; Conway, B.; Tyndall, M. Factors Associated with Spontaneous Clearance of Hepatitis C Virus among Illicit Drug Users. Can. J. Gastroenterol. 2007, 21, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.O.; Adema, K.W.; Bakker, M.; Jurriaans, S.; Berkhout, B.; Cornelissen, M.; Lukashov, V.V. Highly Sensitive Methods Based on Seminested Real-Time Reverse Transcription-PCR for Quantitation of Human Immunodeficiency Virus Type 1 Unspliced and Multiply Spliced RNA and Proviral DNA. J. Clin Microbiol. 2008, 46, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Wong, J.K.; Russenberger, D.; Joos, B.; Opravil, M.; Hirschel, B.; Trkola, A.; Kuster, H.; Weber, R.; Günthard, H.F.; et al. Residual cell-associated unspliced HIV-1 RNA in peripheral blood of patients on potent antiretroviral therapy represents intracellular transcripts. Antivir. Ther. 2002, 7, 91–103. [Google Scholar] [PubMed]
- Emiliani, S.; Van Lint, C.; Fischle, W.; Paras, P.; Ott, M.; Brady, J.; Verdin, E. A point mutation in the HIV-1 Tat responsive element is associated with postintegration latency. Proc. Natl. Acad. Sci. USA 1996, 93, 6377–6381. [Google Scholar] [CrossRef]
- Tabarrini, O.; Massari, S. Editorial: Acting on Tat-Mediated Transcription to Achieve a Long Term Control of HIV-1 Latency. Curr. Pharm. Des. 2017, 23, 4077–4078. [Google Scholar] [CrossRef]
- Kamori, D.; Ueno, T. HIV-1 Tat and Viral Latency: What We Can Learn from Naturally Occurring Sequence Variations. Front. Microbiol. 2017, 8, 80. [Google Scholar] [CrossRef]
- Donahue, D.; Bastarache, S.; Sloan, R.; Wainberg, M. Latent HIV-1 can be reactivated by superinfection in a Tat-dependent manner, which can lead to the emergence of recombinant viruses. Retrovirology 2013, 10 (Suppl. 1), 27. [Google Scholar] [CrossRef]
- Massari, S.; Sabatini, S.; Tabarrini, O. Blocking HIV-1 Replication by Targeting the Tat-Hijacked Transcriptional Machinery. Curr. Pharm. Des. 2013, 19, 1860–1879. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. JAIDS. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Garrido, C.; Spivak, A.M.; Soriano-Sarabia, N.; Checkley, M.A.; Barker, E.; Karn, J.; Planelles, V.; Margolis, D.M. HIV Latency-Reversing Agents Have Diverse Effects on Natural Killer Cell Function. Front. Immunol. 2016, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Zhyvoloup, A.; Melamed, A.; Anderson, I.; Planas, D.; Lee, C.H.; Kriston-Vizi, J.; Ketteler, R.; Merritt, A.; Routy, J.P.; Ancuta, P.; et al. Digoxin reveals a functional connection between HIV-1 integration preference and T-cell activation. PLoS Pathog. 2017, 13, e1006460. [Google Scholar] [CrossRef] [PubMed]
- Schröder, A.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Marini, A.; Harper, J.; Romerio, F. An In Vitro System to Model the Establishment and Reactivation of HIV-1 Latency. J. Immunol. 2008, 181, 7713–7720. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 115) | HIV+ (n = 39) | HIV+/HCV− (n = 34) | HIV+/HCV+ (n = 42) | p | |
|---|---|---|---|---|---|
| Male,n (%) | 69 (60) | 21 (53.8) | 25 (73.5) | 23 (54.8) | 0.158 |
| Median Age, years [median (IQR)] | 50 (43–54) | 47 (39–55) | 51 (43–54.25) | 50 (45–54) | 0.271 |
| Height, cm [median (IQR)] | 165 (158–174) | 164 (154–174) | 168 (160–175) | 164 (160–174) | 0.363 |
| Weight, kg [median (IQR)] | 67.40 (59–79) | 71.50 (61–81) | 77.30 (65–84) | 60 (54–70) | 0.000 |
| Transmission route,n (%) | 0.000 | ||||
| IDUs | 44 (38.3) | 0 (0) | 20 (58.8) | 24 (57.1) | |
| MSM | 30 (26.1) | 15 (38.5) | 9 (26.5) | 6 (14.3) | |
| MSW | 22 (19.1) | 16 (41) | 3 (8.8) | 3 (7.1) | |
| Others | 19 (16.5) | 8 (20.6) | 2 (5.8) | 9 (21.5) | |
| Time of HIV infection, years [median (IQR)] | 18.3 (7.8–26.4) | 14.8 (5.7–25.3) | 20.5 (4.4–26.7) | 19.5 (14.6–28.2) | 0.102 |
| CDC category, n (%) | 0.079 | ||||
| A | 60 (52.8) | 22 (58.4) | 18 (52.9) | 20 (47.6) | |
| B | 17 (14.8) | 5 (12.9) | 4 (11.7) | 8 (19.1) | |
| C | 28 (24.6) | 5 (12.9) | 10 (29.5) | 13 (30.9) | |
| Unknown | 9 (7.8) | 6 (15.4) | 2 (5.9) | 1 (2.4) | |
| ALT | 26.5 (21.4–37.5) | 24.5 (20.0–33.5) | 23.0 (19.0–26.0) | 38.0 (29.0–49.0) | 0.000 |
| AST | 29.0 (21.5–46.5) | 29.5 (22.3–43.8) | 22.0 (12.0–26.6) | 46.0 (31.0–60.5) | 0.000 |
| CMV | 0.671 | ||||
| Yes | 43 (37.4%) | 12 (30.8%) | 14 (41.2%) | 17 (40.5%) | |
| No | 13 (11.3%) | 5 (12.8%) | 5 (14.7%) | 3 (7.1%) | |
| Unknown | 59 (51.3%) | 22 (56.4%) | 15 (44.1%) | 22 (52.4%) |
| Total (n = 115) | HIV+ (n = 39) | HIV+/HCV− (n = 34) | HIV+/HCV+ (n = 42) | p | |
|---|---|---|---|---|---|
| CD4+ T-lymphocytes, cells/µL [median (IQR)] | 763 (636–1004) | 804 (658–1038) | 762 (597–938) | 729 (632–1040) | 0.492 |
| CD4+ T-lymphocytes, % [median (IQR)] | 36 (31–42) | 38 (32–44) | 35 (31–38) | 35.83 (31–42) | 0.299 |
| Nadir CD4, cells/µL [median (IQR)] | 240 (170–338) | 268 (191–407) | 210 (85–299) | 254 (171–336) | 0.224 |
| Nadir CD4 % [median (IQR)] | 18 (10–28) | 24 (17–30) | 13 (6–19) | 21 (13–31) | 0.001 |
| CD8+ T-lymphocytes, cells/µL [median (IQR)] | 863 (639.5–1234) | 845 (539–1437) | 984 (688–1346) | 846 (687–1142) | 0.769 |
| CD8+ T-lymphocytes, % [median (IQR)] | 42 (35.50–45) | 28 (21–46) | 41 (37–45) | 42.50 (41–47) | 0.090 |
| CD4:CD8 Ratio | 0.9 (0.7–1.1) | 1.0 (0.8–1.4) | 0.9 (0.7–1.1) | 0.9 (0.7–1.1) | 0.087 |
| ART regimen, n (%) | 0.889 | ||||
| NNRTIs | 40 (34.8) | 11 (28.8) | 14 (41.2) | 15 (35.7) | |
| NRTIs | 1 (0.9) | 0 (0) | 1 (2.9) | 0 (0) | |
| PIs | 12 (10.4) | 3 (7.7) | 5 (14.7) | 4 (9.5) | |
| INIs | 42 (36.5) | 17 (43.6) | 10 (29.4) | 15 (35.7) | |
| Dual Therapy | 14 (12.2) | 5 (12.8) | 3 (8.8) | 6 (14.3) | |
| Monotherapy | 5 (4.3) | 2 (5.1) | 1 (2.9) | 2 (4.8) | |
| Unknown | 1 (0.9) | 1 (0.9) | 0 (0) | 0 (0) |
| Total (n = 115) | HIV+/HCV− (n = 34) | HIV+/HCV+ (n = 42) | p | |
|---|---|---|---|---|
| Fibrosis,n (%) | 0.000 | |||
| F0–F1 (< 6 kPA) | 55 (72.4) | 23 (67.6) | 29 (69) | |
| F2 (6–9 kPa) | 7 (6.1) | 0 (0) | 7 (16.7) | |
| F3 (> 9–12 kPa) | 3 (2.6) | 0 (0) | 3 (7.1) | |
| F4 (> 12 kPa) | 1 (0.9) | 0 (0) | 1 (2.4) | |
| Unknown | 13 (17.1) | 11 (32.4) | 2 (4.8) | |
| HCV genotype, n (%) | 0.000 | |||
| 1 | 26 (34.2) | 1 (2.9) | 25 (59.5) | |
| 2 | 2 (2.6) | 0 (0) | 2 (4.8) | |
| 3 | 4 (5.26) | 0 (0) | 4 (9.5) | |
| 4 | 11 (14.5) | 2 (5.9) | 8 (19) | |
| Unknown | 34 (44.7) | 31 (91.2) | 3 (7.1) | |
| IL-28 genotype,n (%) | 0.021 | |||
| CC | 37 (48.7) | 23 (67.6) | 14 (33.3) | |
| CT/TC | 30 (39.5) | 9 (26.5) | 21 (50) | |
| TT | 6 (7.9) | 2 (5.9) | 4 (9.5) | |
| Unknown | 3 (3.9) | 0 (0) | 3 (7.1) |
| Groups | Primary Outcome Mean ± SEM | Univariate Analysis ARM (95% CI) | p | Multivariate Analysis aAMR (95% CI) | p |
|---|---|---|---|---|---|
| HIV+ | 60.14 ± 11.28 | 0 | - | 0 | - |
| HIV+/HCV− | 100.60 ± 19.49 | 1.673 (0.986; 2.838) | 0.056 | 1.815 (1.050; 3.138) | 0.033 |
| HIV+/HCV+ | 102.88 ± 18.20 | 1.711 (1.032; 2.836) | 0.037 | 1.684 (1.018; 2.785) | 0.042 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Román, P.; López-Huertas, M.R.; Crespo-Bermejo, C.; Arca-Lafuente, S.; Cortegano, I.; Valle-Millares, D.; Gaspar, M.L.; Martín-Carbonero, L.; Domínguez-Domínguez, L.; Ryan, P.; et al. Hepatitis C Virus Influences HIV-1 Viral Splicing in Coinfected Patients. J. Clin. Med. 2020, 9, 2091. https://doi.org/10.3390/jcm9072091
Martínez-Román P, López-Huertas MR, Crespo-Bermejo C, Arca-Lafuente S, Cortegano I, Valle-Millares D, Gaspar ML, Martín-Carbonero L, Domínguez-Domínguez L, Ryan P, et al. Hepatitis C Virus Influences HIV-1 Viral Splicing in Coinfected Patients. Journal of Clinical Medicine. 2020; 9(7):2091. https://doi.org/10.3390/jcm9072091
Chicago/Turabian StyleMartínez-Román, Paula, María Rosa López-Huertas, Celia Crespo-Bermejo, Sonia Arca-Lafuente, Isabel Cortegano, Daniel Valle-Millares, María Luisa Gaspar, Luz Martín-Carbonero, Lourdes Domínguez-Domínguez, Pablo Ryan, and et al. 2020. "Hepatitis C Virus Influences HIV-1 Viral Splicing in Coinfected Patients" Journal of Clinical Medicine 9, no. 7: 2091. https://doi.org/10.3390/jcm9072091
APA StyleMartínez-Román, P., López-Huertas, M. R., Crespo-Bermejo, C., Arca-Lafuente, S., Cortegano, I., Valle-Millares, D., Gaspar, M. L., Martín-Carbonero, L., Domínguez-Domínguez, L., Ryan, P., de los Santos, I., de la Fuente-Moral, S., Fernández-Rodríguez, A., Coiras, M., & Briz, V., on behalf of COVIHEP. (2020). Hepatitis C Virus Influences HIV-1 Viral Splicing in Coinfected Patients. Journal of Clinical Medicine, 9(7), 2091. https://doi.org/10.3390/jcm9072091