Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far?

 , ,
, ,
Abstract
:1. Introduction
1.1. Why Gene Therapy?
1.2. A Brief History of Gene Therapy
1.3. Gene Therapy Challenges in Rare Respiratory Diseases
2. Gene Therapy Strategies
2.1. Transfection Vectors
2.2. Viral Vectors
2.3. Non Viral Vectors
2.4. Physical Methods
2.5. Inorganic Particles
2.6. Biocomponents
2.7. Gene Editing Techniques
2.8. Zinc Fingers and Transcription Activator-Like Effector Nucleases
2.9. CRISPR/Cas9
2.10. Cellular and Animal Models
2.11. Animal Models in CF
2.12. Animal Models in AATD
2.13. Animal Models in PCD
3. Gene Therapy in Rare Respiratory Diseases
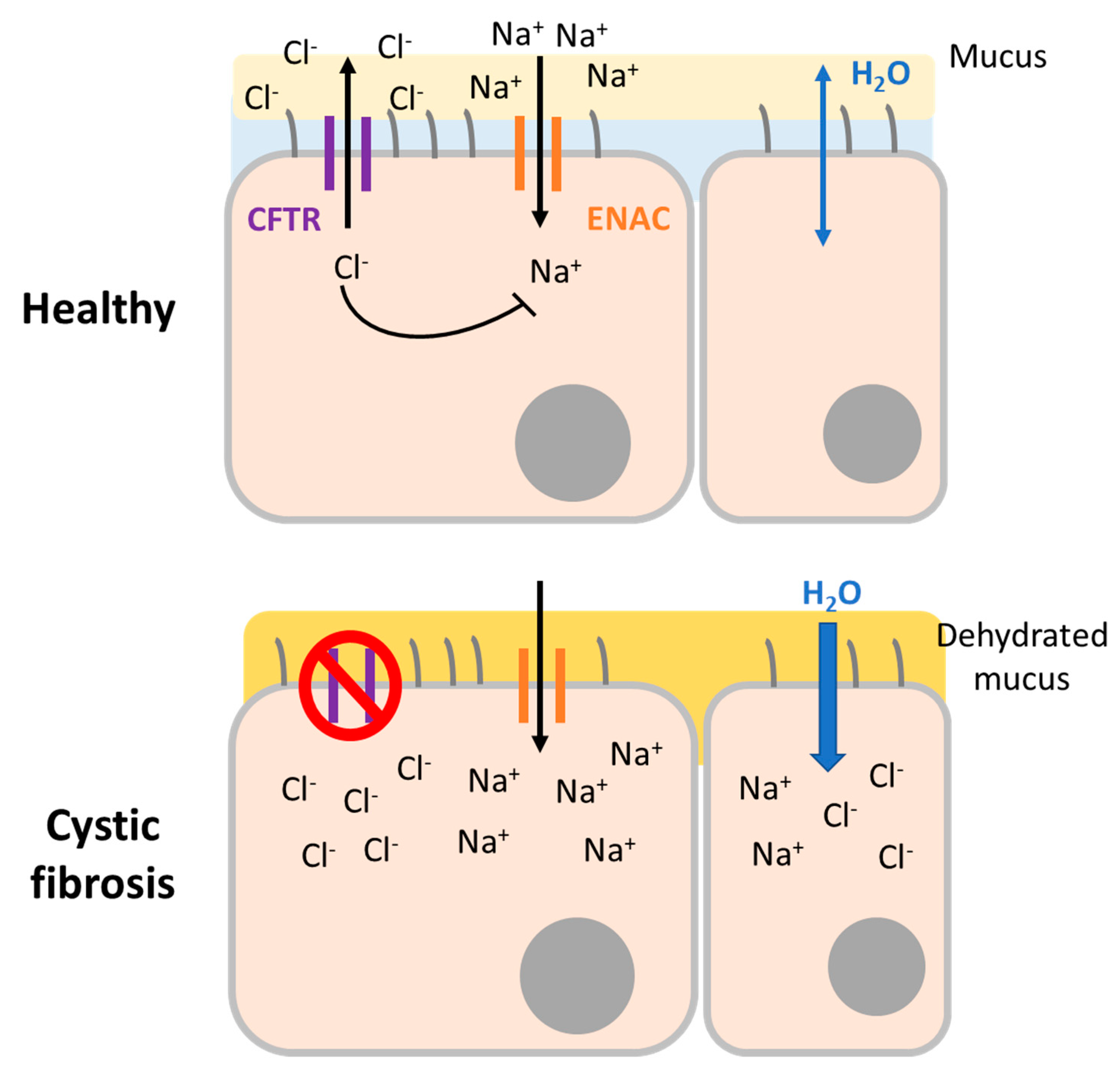
3.1. Cystic Fibrosis
3.1.1. Adenoviral Vectors
3.1.2. Lentiviral Vectors
3.1.3. Recombinant Adeno-Associated Viral Vectors
3.1.4. Non-Viral Vectors
3.1.5. CFTR Gene Correction
3.1.6. Conclusion
3.2. Alpha-1 Antitrypsin Deficiency, the Genetic COPD
3.2.1. Viral Gene Augmentation Therapy
3.2.2. Dual Therapy Approach: Addressing Hepatic and Respiratory Disease with MiRNA
3.2.3. Non-Viral Therapy
3.3. SERPINA1 Gene Correction
Conclusions
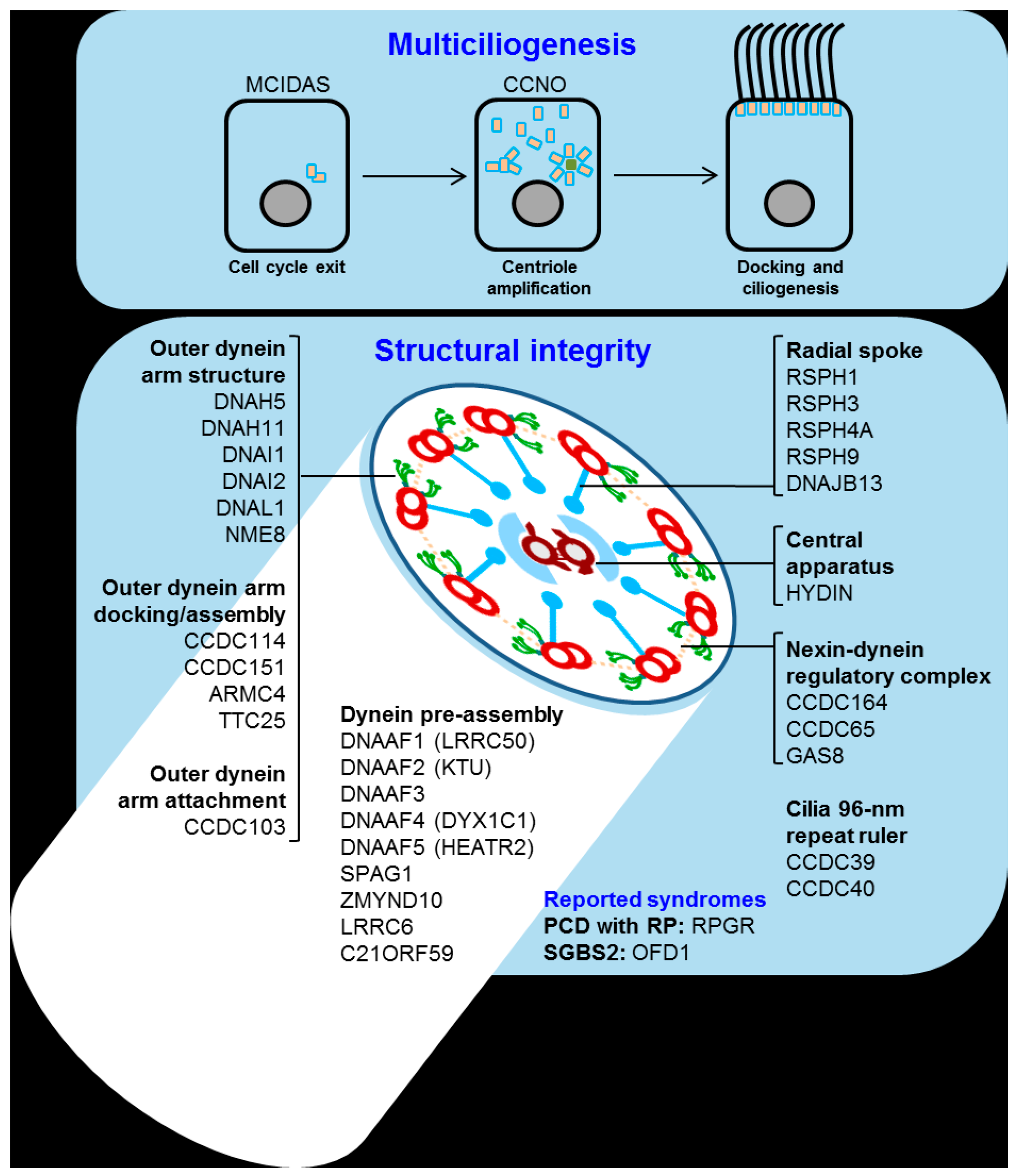
4. Primary Ciliary Dyskinesia
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xiong, F.; Mi, Z.; Gu, N.; Gu, N. Cationic liposomes as gene delivery system: Transfection efficiency and new application. Pharmazie 2011, 66, 158–164. [Google Scholar] [CrossRef]
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2009, 96, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet Lond. Engl. 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Sheridan, C. Go-ahead for first in-body CRISPR medicine testing. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Single Ascending Dose Study in Participants with LCA10-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03872479 (accessed on 18 June 2020).
- Aitken, M.L.; Moss, R.B.; Waltz, D.A.; Dovey, M.E.; Tonelli, M.R.; McNamara, S.C.; Gibson, R.L.; Ramsey, B.W.; Carter, B.J.; Reynolds, T.C. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum. Gene Ther. 2001, 12, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Sullivan, K.; Wetzel, R.; Taylor, G.; Carter, B.J.; Guggino, W.B.; Zeitlin, P.L.; Reynolds, T.C.; Heald, A.E.; Pedersen, P.; et al. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (raav2)-cftr vector in adult cystic fibrosis patients: A two-part clinical study. Hum. Gene Ther. 2003, 14, 1079–1088. [Google Scholar] [CrossRef]
- Stiles, K.M.; Sondhi, D.; Kaminsky, S.M.; De, B.P.; Rosenberg, J.B.; Crystal, R.G. Intrapleural Gene Therapy for Alpha-1 Antitrypsin Deficiency-Related Lung Disease. Chronic Obstr. Pulm. Dis. Miami Fla 2018, 5, 244–257. [Google Scholar] [CrossRef]
- Pickles, R.J. Physical and biological barriers to viral vector-mediated delivery of genes to the airway epithelium. Proc. Am. Thorac. Soc. 2004, 1, 302–308. [Google Scholar] [CrossRef]
- Kolb, M.; Martin, G.; Medina, M.; Ask, K.; Gauldie, J. Gene therapy for pulmonary diseases. Chest 2006, 130, 879–884. [Google Scholar] [CrossRef]
- Ferrari, S.; Griesenbach, U.; Geddes, D.M.; Alton, E. Immunological hurdles to lung gene therapy. Clin. Exp. Immunol. 2003, 132, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G.; Randell, S.H.; Engelhardt, J.F.; Voynow, J.; Sunday, M.E. Airway epithelial cells: Current concepts and challenges. Proc. Am. Thorac. Soc. 2008, 5, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, D.; Stiles, K.M.; De, B.P.; Crystal, R.G. Genetic modification of the lung directed toward treatment of human disease. Hum. Gene Ther. 2017, 28, 3–84. [Google Scholar] [CrossRef] [PubMed]
- Baliou, S.; Adamaki, M.; Kyriakopoulos, A.M.; Spandidos, D.A.; Panayiotidis, M.; Christodoulou, I.; Zoumpourlis, V. CRISPR therapeutic tools for complex genetic disorders and cancer (Review). Int. J. Oncol. 2018, 53, 443–468. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Marquez Loza, L.I.; Yuen, E.C.; McCray, P.B. Lentiviral Vectors for the Treatment and Prevention of Cystic Fibrosis Lung Disease. Genes 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, J.; Wandtke, T.; Kopinski, P.; Chorostowska-Wynimko, J. Challenges and Prospects for Alpha-1 Antitrypsin Deficiency Gene Therapy. Hum. Gene Ther. 2015, 26, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, L.E.; Yin, W.; Patel, M.; Sechelski, J.; Rogers, T.; Burns, K.; Grubb, B.R.; Olsen, J.C. Restoring ciliary function to differentiated primary ciliary dyskinesia cells with a lentiviral vector. Gene Ther. 2014, 21, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Cooney, A.L.; McCray, P.B.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; McCray Jr, P.B.; Engelhardt, J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019, 28, R88–R94. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, M.A.; Siegfried, W.; Yoshimura, K.; Yoneyama, K.; Fukayama, M.; Stier, L.E.; Pääkkö, P.K.; Gilardi, P.; Stratford-Perricaudet, L.D.; Perricaudet, M. Adenovirus-mediated transfer of a recombinant alpha 1-antitrypsin gene to the lung epithelium in vivo. Science 1991, 252, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Chulay, J.D.; Ye, G.-J.; Thomas, D.L.; Knop, D.R.; Benson, J.M.; Hutt, J.A.; Wang, G.; Humphries, M.; Flotte, T.R. Preclinical Evaluation of a Recombinant Adeno-Associated Virus Vector Expressing Human Alpha-1 Antitrypsin Made Using a Recombinant Herpes Simplex Virus Production Method. Hum. Gene Ther. 2011, 22, 155–165. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.M.; Wells, K.E.; Bamfo, J.E.; Cartwright, M.A.; Wells, D.J. Inflammatory responses following direct injection of plasmid DNA into skeletal muscle. Gene Ther. 1998, 5, 1283–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, W.; Ma, N.; Steinhoff, G. Non-Viral Gene Delivery Methods. Curr. Pharm. Biotechnol. 2013, 14, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Eberwine, J.H. Mammalian cell transfection: The present and the future. Anal. Bioanal. Chem. 2010, 397, 3173–3178. [Google Scholar] [CrossRef] [Green Version]
- DeMayo, J.L.; Wang, J.; Liang, D.; Zhang, R.; DeMayo, F.J. Genetically Engineered Mice by Pronuclear DNA microinjection. Curr. Protoc. Mouse Biol. 2012, 2, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Herrero, M.J.; Sendra, L.; Miguel, A.; Aliño, S.F. Physical Methods of Gene Delivery. In Safety and Efficacy of Gene-Based Therapeutics for Inherited Disorders; Brunetti-Pierri, N., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 113–115. ISBN 978-3-319-53457-2. [Google Scholar]
- Sherba, J.J.; Hogquist, S.; Lin, H.; Shan, J.W.; Shreiber, D.I.; Zahn, J.D. The effects of electroporation buffer composition on cell viability and electro-transfection efficiency. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mellott, A.J.; Forrest, M.L.; Detamore, M.S. Physical non-viral gene delivery methods for tissue engineering. Ann. Biomed. Eng. 2013, 41, 446–468. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lei, J.; Vollmer, R.; Huang, L. Mechanism of Liver Gene Transfer by Mechanical Massage. Mol. Ther. 2004, 9, 452–457. [Google Scholar] [CrossRef]
- Giner-Casares, J.J.; Henriksen-Lacey, M.; Coronado-Puchau, M.; Liz-Marzán, L.M. Inorganic nanoparticles for biomedicine: Where materials scientists meet medical research. Mater. Today 2016, 19, 19–28. [Google Scholar] [CrossRef]
- Guo, L.; Wang, L.; Yang, R.; Feng, R.; Li, Z.; Zhou, X.; Dong, Z.; Ghartey-Kwansah, G.; Xu, M.; Nishi, M.; et al. Optimizing conditions for calcium phosphate mediated transient transfection. Saudi J. Biol. Sci. 2017, 24, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lou, C.; Yang, H.; Shi, M.; Miyoshi, H. Silica nanoparticles as promising drug/gene delivery carriers and fluorescent nano-probes: Recent advances. Curr. Cancer Drug Targets 2011, 11, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Huefner, A.; Septiadi, D.; Wilts, B.D.; Patel, I.I.; Kuan, W.L.; Fragniere, A.; Barker, R.A.; Mahajan, S. Gold nanoparticles explore cells: Cellular uptake and their use as intracellular probes. Methods 2014, 68, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, A.; Claypool, S.; Liu, R. The Smart Targeting of Nanoparticles. Curr. Pharm. Des. 2013, 19, 6315–6329. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.; Meng, Q.; Liu, K. Peptide-based gene delivery vectors. J. Mater. Chem. B 2019, 7, 1824–1841. [Google Scholar] [CrossRef]
- Baker, A.; Saltik, M.; Lehrmann, H.; Killisch, I.; Mautner, V.; Lamm, G.; Christofori, G.; Cotten, M. Polyethylenimine (PEI) is a simple, inexpensive and effective reagent for condensing and linking plasmid DNA to adenovirus for gene delivery. Gene Ther. 1997, 4, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Santos-Carballal, B.; Fernández, E.F.; Goycoolea, F.M. Chitosan in non-viral gene delivery: Role of structure, characterization methods, and insights in cancer and rare diseases therapies. Polymers 2018, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, M.; Esenaliev, R. PLGA Nanoparticles for Ultrasound-Mediated Gene Delivery to Solid Tumors. J. Drug Deliv. 2012, 2012. [Google Scholar] [CrossRef]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Mol. J. Synth. Chem. Nat. Prod. Chem. 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Christiaens, B.; Dubruel, P.; Grooten, J.; Goethals, M.; Vandekerckhove, J.; Schacht, E.; Rosseneu, M. Enhancement of polymethacrylate-mediated gene delivery by Penetratin. Eur. J. Pharm. Sci. 2005, 24, 525–537. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Mayerl, S.J.; Chan, A.H.; Conklin, B.R. Detection and Quantification of HDR and NHEJ Induced by Genome Editing at Endogenous Gene Loci Using Droplet Digital PCR. Methods Mol. Biol. Clifton NJ 2018, 1768, 349–362. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [Green Version]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Keith Joung, J. CRISPR RNA–guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Gajula, K.S. Designing an Elusive C•G→G•C CRISPR Base Editor. Trends Biochem. Sci. 2019, 44, 91–94. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Gopalappa, R.; Suresh, B.; Ramakrishna, S.; Kim, H. Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res. 2018, 46, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Cox, D.B.T.; Yan, W.; Manteiga, J.; Schneider, M.; Yamano, T.; Nishimasu, H.; Nureki, O.; Zhang, F. Engineered Cpf1 Enzymes with Altered PAM Specificities. bioRxiv 2016, 091611–091611. [Google Scholar] [CrossRef]
- Torres-Durán, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef] [Green Version]
- Meurens, F.; Summerfield, A.; Nauwynck, H.; Saif, L.; Gerdts, V. The pig: A model for human infectious diseases. Trends Microbiol. 2012, 20, 50–57. [Google Scholar] [CrossRef]
- Ostedgaard, L.S.; Rogers, C.S.; Dong, Q.; Randak, C.O.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Welsh, M.J. Processing and function of CFTR-DeltaF508 are species-dependent. Proc. Natl. Acad. Sci. USA 2007, 104, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.T.; Liu, X.; Yan, Z.; Luo, M.; Zhang, Y.; Zhou, W.; Lee, B.J.; Song, Y.; Guo, C.; Wang, Y.; et al. Comparative Processing and Function of Human and Ferret Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 2012, 287, 21673–21685. [Google Scholar] [CrossRef] [Green Version]
- Harris, A. Towards an ovine model of cystic fibrosis. Hum. Mol. Genet. 1997, 6, 2191–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tata, F.; Stanier, P.; Wicking, C.; Halford, S.; Kruyer, H.; Lench, N.J.; Scambler, P.J.; Hansen, C.; Braman, J.C.; Williamson, R. Cloning the mouse homolog of the human cystic fibrosis transmembrane conductance regulator gene. Genomics 1991, 10, 301–307. [Google Scholar] [CrossRef]
- Trezise, A.E.; Szpirer, C.; Buchwald, M. Localization of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) in the rat to chromosome 4 and implications for the evolution of mammalian chromosomes. Genomics 1992, 14, 869–874. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [Green Version]
- Semaniakou, A.; Croll, R.P.; Chappe, V. Animal Models in the Pathophysiology of Cystic Fibrosis. Front. Pharmacol. 2018, 9, 1475. [Google Scholar] [CrossRef] [Green Version]
- Ghorani, V.; Boskabady, M.H.; Khazdair, M.R.; Kianmeher, M. Experimental animal models for COPD: A methodological review. Tob. Induc. Dis. 2017, 15. [Google Scholar] [CrossRef]
- Sifers, R.N.; Carlson, J.A.; Clift, S.M.; DeMayo, F.J.; Bullock, D.W.; Woo, S.L.C. Tissue specific expression of the human alpha-1-antitrypsin gene in transgenic mice. Nucleic Acids Res. 1987, 15, 1459–1475. [Google Scholar] [CrossRef] [Green Version]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Finegold, M.J.; Clift, S.M.; DeMayo, F.J.; Bullock, D.W.; Woo, S.L. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Investig. 1989, 83, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Borel, F.; Sun, H.; Zieger, M.; Cox, A.; Cardozo, B.; Li, W.; Oliveira, G.; Davis, A.; Gruntman, A.; Flotte, T.R.; et al. Editing out five Serpina1 paralogs to create a mouse model of genetic emphysema. Proc. Natl. Acad. Sci. USA 2018, 115, 2788–2793. [Google Scholar] [CrossRef] [Green Version]
- Ni, K.; Serban, K.A.; Batra, C.; Petrache, I. Alpha-1 Antitrypsin Investigations Using Animal Models of Emphysema. Ann. Am. Thorac. Soc. 2016, 13, S311–S316. [Google Scholar] [CrossRef]
- Eggenschwiler, R.; Patronov, A.; Hegermann, J.; Fráguas-Eggenschwiler, M.; Wu, G.; Cortnumme, L.; Ochs, M.; Antes, I.; Cantz, T. A combined in silico and in vitro study on mouse Serpina1a antitrypsin-deficiency mutants. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yip, E.; Giousoh, A.; Fung, C.; Wilding, B.; Prakash, M.D.; Williams, C.; Verkade, H.; Bryson-Richardson, R.J.; Bird, P.I. A transgenic zebrafish model of hepatocyte function in human Z α1-antitrypsin deficiency. Biol. Chem. 2019, 400, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Norris, D.P.; Grimes, D.T. Mouse models of ciliopathies: The state of the art. Dis. Model. Mech. 2012, 5, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirschell, M.; Yamamoto, R.; Alford, L.; Gokhale, A.; Gaillard, A.; Sale, W.S. Regulation of ciliary motility: Conserved protein kinases and phosphatases are targeted and anchored in the ciliary axoneme. Arch. Biochem. Biophys. 2011, 510, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Schmid, A.; Salathe, M. Ciliary beat co-ordination by calcium. Biol. Cell 2011, 103, 159–169. [Google Scholar] [CrossRef]
- Dean, S.; Sunter, J.D.; Wheeler, R.J. TrypTag.org: A Trypanosome Genome-wide Protein Localisation Resource. Trends Parasitol. 2017, 33, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Dubruille, R.; Laurençon, A.; Vandaele, C.; Shishido, E.; Coulon-Bublex, M.; Swoboda, P.; Couble, P.; Kernan, M.; Durand, B. Drosophila regulatory factor X is necessary for ciliated sensory neuron differentiation. Development 2002, 129, 5487–5498. [Google Scholar] [CrossRef] [Green Version]
- Marshall, W.F.; Kintner, C. Cilia orientation and the fluid mechanics of development. Curr. Opin. Cell Biol. 2008, 20, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Schweickert, A.; Vick, P.; Wright, C.V.E.; Danilchik, M.V. Symmetry breakage in the vertebrate embryo: When does it happen and how does it work? Dev. Biol. 2014, 393, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Zhao, L.; Brueckner, M.; Sun, Z. Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr. Biol. 2015, 25, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Brennan, M.-L.; Schrijver, I. Cystic Fibrosis: A Review of Associated Phenotypes, Use of Molecular Diagnostic Approaches, Genetic Characteristics, Progress, and Dilemmas. J. Mol. Diagn. JMD 2016, 18, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, N.A. CFTR and Cystic Fibrosis: A Need for Personalized Medicine. In Ion Channels and Transporters of Epithelia in Health and Disease; Springer New York: New York, NY, USA, 2016; pp. 773–802. [Google Scholar]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M.J. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Crystal, R.G.; McElvaney, N.G.; Rosenfeld, M.A.; Chu, C.S.; Mastrangeli, A.; Hay, J.G.; Brody, S.L.; Jaffe, H.A.; Eissa, N.T.; Danel, C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet. 1994, 8, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Ramsey, B.W.; Meeker, D.P.; Aitken, M.L.; Balfour, R.P.; Gibson, R.L.; Launspach, J.; Moscicki, R.A.; Richards, S.M.; Standaert, T.A.; et al. Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J. Clin. Investig. 1996, 97, 1504–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Ouyang, H.; Grasemann, H.; Bartlett, C.; Du, K.; Duan, R.; Shi, F.; Estrada, M.; Seigel, K.E.; Coates, A.L.; et al. Transducing Airway Basal Cells with a Helper-Dependent Adenoviral Vector for Lung Gene Therapy. Hum. Gene Ther. 2018, 29, 643–652. [Google Scholar] [CrossRef]
- Mitomo, K.; Griesenbach, U.; Inoue, M.; Somerton, L.; Meng, C.; Akiba, E.; Tabata, T.; Ueda, Y.; Frankel, G.M.; Farley, R.; et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with sendai virus envelopes. Mol. Ther. 2010, 18, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Alton, E.W.F.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Flotte, T.; Carter, B.; Conrad, C.; Guggino, W.; Reynolds, T.; Rosenstein, B.; Taylor, G.; Walden, S.; Wetzel, R. A phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung disease. Hum. Gene Ther. 1996, 7, 1145–1159. [Google Scholar] [CrossRef]
- Wagner, J.A.; Reynolds, T.; Moran, M.L.; Moss, R.B.; Wine, J.J.; Flotte, T.R.; Gardner, P. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet 1998, 351, 1702–1703. [Google Scholar] [CrossRef]
- Wagner, J.A.; Nepomuceno, I.B.; Messner, A.H.; Moran, M.L.; Batson, E.P.; Dimiceli, S.; Brown, B.W.; Desch, J.K.; Norbash, A.M.; Conrad, C.K.; et al. A Phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther. 2002, 13, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; Rodman, D.; Spencer, L.T.; Aitken, M.L.; Zeitlin, P.L.; Waltz, D.; Milla, C.; Brody, A.S.; Clancy, J.P.; Ramsey, B.; et al. Repeated Adeno-Associated Virus Serotype 2 Aerosol-Mediated Cystic Fibrosis Transmembrane Regulator Gene Transfer to the Lungs of Patients with Cystic Fibrosis: A Multicenter, Double-Blind, Placebo-Controlled Trial. Chest 2004, 125, 509–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, R.B.; Milla, C.; Colombo, J.; Accurso, F.; Zeitlin, P.L.; Clancy, J.P.; Spencer, L.T.; Pilewski, J.; Waltz, D.A.; Dorkin, H.L.; et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: A randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007, 18, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.C.; Smith, C.I.; Cebotaru, L.; Zhang, X.; Askin, F.B.; Wright, J.; Guggino, S.E.; Adams, R.J.; Flotte, T.; Guggino, W.B. Expression of a truncated cystic Fibrosis transmembrane conductance regulator with an AAV5-pseudotyped vector in primates. Mol. Ther. 2007, 15, 756–763. [Google Scholar] [CrossRef]
- Flotte, T.R.; Fischer, A.C.; Goetzmann, J.; Mueller, C.; Cebotaru, L.; Yan, Z.; Wang, L.; Wilson, J.M.; Guggino, W.B.; Engelhardt, J.F. Dual reporter comparative indexing of rAAV pseudotyped vectors in chimpanzee airway. Mol. Ther. 2010, 18, 594–600. [Google Scholar] [CrossRef]
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Adeno-Associated Virus Type 6 (AAV6) Vectors Mediate Efficient Transduction of Airway Epithelial Cells in Mouse Lungs Compared to That of AAV2 Vectors. J. Virol. 2001, 75, 6615–6624. [Google Scholar] [CrossRef] [Green Version]
- Excoffon, K.J.D.A.; Koerber, J.T.; Dickey, D.D.; Murtha, M.; Keshavjee, S.; Kaspar, B.K.; Zabner, J.; Schaffer, D.V. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3865–3870. [Google Scholar] [CrossRef] [Green Version]
- Steines, B.; Dickey, D.D.; Bergen, J.; Excoffon, K.J.D.A.; Weinstein, J.R.; Li, X.; Yan, Z.; Alaiwa, M.H.A.; Shah, V.S.; Bouzek, D.C.; et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight 2016, 1, e88728–e88728. [Google Scholar] [CrossRef]
- Alton, E.W.F.W.; Boyd, A.C.; Porteous, D.J.; Davies, G.; Davies, J.C.; Griesenbach, U.; Higgins, T.E.; Gill, D.R.; Hyde, S.C.; Innes, J.A.; et al. A Phase I/IIa Safety and Efficacy Study of Nebulized Liposome-mediated Gene Therapy for Cystic Fibrosis Supports a Multidose Trial. Am. J. Respir. Crit. Care Med. 2015, 192, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Alton, E.W.F.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; Davis, P.B.; Wagener, J.S.; Hilliard, K.A.; Stern, R.C.; Milgram, L.J.H.; Kowalczyk, T.H.; Hyatt, S.L.; Fink, T.L.; Gedeon, C.R.; et al. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum. Gene Ther. 2004, 15, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Flynn, R.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Correction of the Δf508 mutation in the cystic fibrosis transmembrane conductance regulator gene by zinc-finger nuclease homology-directed repair. BioResearch Open Access 2012, 1, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Sargent, R.G.; Illek, B.; Fischer, H.; Esmaeili-Shandiz, A.; Yezzi, M.J.; Lee, A.; Yang, Y.; Kim, S.; Renz, P.; et al. TALENs facilitate single-step seamless SDF correction of F508del CFTR in airway epithelial submucosal gland cell-derived CF-iPSCs. Mol. Ther. Nucleic Acids 2016, 5, e273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkert, S.; Bednarski, C.; Göhring, G.; Cathomen, T.; Martin, U. Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Res. 2017, 23, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.; Zhang, Y.; Cao, H.; Li, J.; Duan, R.; Hu, J. TALEN-Mediated Gene Targeting for Cystic Fibrosis-Gene Therapy. Genes 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; Van Der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Hirai, H.; Yang, D.; Ma, L.; Hou, X.; Jiang, H.; Wei, H.; Rajagopalan, C.; Mou, H.; Wang, G.; et al. Efficient Gene Editing at Major CFTR Mutation Loci. Mol. Ther. Nucleic Acids 2019, 16, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017, 12, e0184009. [Google Scholar] [CrossRef] [Green Version]
- De Serres, F.J.; Blanco, I. Prevalence of α1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: A comprehensive review. Ther. Adv. Respir. Dis. 2012, 6, 277–295. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.M.; Kim, Y.; Shim, J.S.; Park, J.T.; Wang, R.H.; Leach, S.D.; Liu, J.O.; Deng, C.; Ye, Z.; Jang, Y.Y. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 2013, 57, 2458–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Morgan, M.; Ellis, T.; Poirier, A.; Chesnut, K.; Wang, J.; Brantly, M.; Muzyczka, N.; Byrne, B.J.; Atkinson, M.; et al. Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectors. Proc. Natl. Acad. Sci. USA 1998, 95, 14384–14388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantly, M.L.; Spencer, L.T.; Humphries, M.; Conlon, T.J.; Spencer, C.T.; Poirier, A.; Garlington, W.; Baker, D.; Song, S.; Berns, K.I.; et al. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 α1-antitrypsin (AAT) vector in AAT-deficient adults. Hum. Gene Ther. 2006, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Brantly, M.L.; Chulay, J.D.; Wang, L.; Mueller, C.; Humphries, M.; Spencer, L.T.; Rouhani, F.; Conlon, T.J.; Calcedo, R.; Betts, M.R.; et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 16363–16368. [Google Scholar] [CrossRef] [Green Version]
- Flotte, T.R.; Trapnell, B.C.; Humphries, M.; Carey, B.; Calcedo, R.; Rouhani, F.; Campbell-Thompson, M.; Yachnis, A.T.; Sandhaus, R.A.; McElvaney, N.G.; et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing α 1-antitrypsin: Interim results. Hum. Gene Ther. 2011, 22, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Mueller, C.; Gernoux, G.; Gruntman, A.M.; Borel, F.; Reeves, E.P.; Calcedo, R.; Rouhani, F.N.; Yachnis, A.; Humphries, M.; Campbell-Thompson, M.; et al. 5 Year Expression and Neutrophil Defect Repair after Gene Therapy in Alpha-1 Antitrypsin Deficiency. Mol. Ther. 2017, 25, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- De, B.; Heguy, A.; Leopold, P.L.; Wasif, N.; Korst, R.J.; Hackett, N.R.; Crystal, R.G. Intrapleural administration of a serotype 5 adeno-associated virus coding for α1-antitrypsin mediates persistent, high lung and serum levels of α1-antitrypsin. Mol. Ther. 2004, 10, 1003–1010. [Google Scholar] [CrossRef]
- De, B.P.; Heguy, A.; Hackett, N.R.; Ferris, B.; Leopold, P.L.; Lee, J.; Pierre, L.; Gao, G.; Wilson, J.M.; Crystal, R.G. High levels of persistent expression of α1-Antitrypsin mediated by the nonhuman primate serotype rh.10 adeno-associated virus despite preexisting immunity to common human adeno-associated viruses. Mol. Ther. 2006, 13, 67–76. [Google Scholar] [CrossRef]
- Stoll, S.M.; Sclimenti, C.R.; Baba, E.J.; Meuse, L.; Kay, M.A.; Calos, M.P. Epstein-Barr virus/human vector provides high-level, long-term expression of α1-antitrypsin in mice. Mol. Ther. 2001, 4, 122–129. [Google Scholar] [CrossRef]
- Wilson, A.A.; Murphy, G.J.; Hamakawa, H.; Kwok, L.W.; Srinivasan, S.; Hovav, A.H.; Mulligan, R.C.; Amar, S.; Suki, B.; Kotton, D.N. Amelioration of emphysema in mice through lentiviral transduction of long-lived pulmonary alveolar macrophages. J. Clin. Investig. 2010, 120, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Liqun Wang, R.; McLaughlin, T.; Cossette, T.; Tang, Q.; Foust, K.; Campbell-Thompson, M.; Martino, A.; Cruz, P.; Loiler, S.; Mueller, C.; et al. Recombinant AAV serotype and capsid mutant comparison for pulmonary gene transfer of α-1-antitrypsin using invasive and noninvasive delivery. Mol. Ther. 2009, 17, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.J.; Kashentseva, E.; Everett, W.; Kaliberova, L.; Curiel, D.T. Targeted in vivo knock-in of human alpha-1-antitrypsin cDNA using adenoviral delivery of CRISPR/Cas9. Gene Ther. 2018, 25, 139–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Lu, Y.; Witek, R.P.; Chang, L.J.; Campbell-Thompson, M.; Jorgensen, M.; Petersen, B.; Song, S. Ex vivo transduction and transplantation of bone marrow cells for liver gene delivery of α1-antitrypsin. Mol. Ther. 2010, 18, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.A.; Kwok, L.W.; Hovav, A.H.; Ohle, S.J.; Little, F.F.; Fine, A.; Kotton, D.N. Sustained expression of α1-antitrypsin after transplantation of manipulated hematopoietic stem cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaedi, M.; Lotfi, A.S.; Soleimani, M. Establishment of lentiviral-vector-mediated model of human alpha-1 antitrypsin delivery into hepatocyte-like cells differentiated from mesenchymal stem cells. Tissue Cell 2010, 42, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Tang, Q.; Gruntman, A.; Blomenkamp, K.; Teckman, J.; Song, L.; Zamore, P.D.; Flotte, T.R. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol. Ther. 2012, 20, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Aliño, S.F.; Crespo, A.; Dasí, F. Long-term therapeutic levels of human alpha-1 antitrypsin in plasma after hydrodynamic injection of nonviral DNA. Gene Ther. 2003, 10, 1672–1679. [Google Scholar] [CrossRef]
- Sendra, L.; Miguel, A.; Pérez-Enguix, D.; Herrero, M.J.; Montalvá, E.; García-Gimeno, M.A.; Noguera, I.; Díaz, A.; Pérez, J.; Sanz, P.; et al. Studying closed hydrodynamic models of “in vivo” DNA perfusion in pig liver for gene therapy translation to humans. PLoS ONE 2016, 11, e0163898. [Google Scholar] [CrossRef]
- Sendra Gisbert, L.; Miguel Matas, A.; Sabater Ortí, L.; Herrero, M.J.; Sabater Olivas, L.; Montalvá Orón, E.M.; Frasson, M.; Abargues López, R.; López-Andújar, R.; García-Granero Ximénez, E.; et al. Efficacy of hydrodynamic interleukin 10 gene transfer in human liver segments with interest in transplantation. Liver Transpl. 2017, 23, 50–62. [Google Scholar] [CrossRef]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.Q.; Paschon, D.E.; Miranda, E.; Ordóñez, A.; Hannan, N.R.F.; Rouhani, F.J.; et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature 2011, 478, 391–394. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Abalde-Atristain, L.; He, C.; Brodsky, B.R.; Braunstein, E.M.; Chaudhari, P.; Jang, Y.Y.; Cheng, L.; Ye, Z. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther. 2015, 23, 570–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjursell, M.; Porritt, M.J.; Ericson, E.; Taheri-Ghahfarokhi, A.; Clausen, M.; Magnusson, L.; Admyre, T.; Nitsch, R.; Mayr, L.; Aasehaug, L.; et al. Therapeutic Genome Editing With CRISPR/Cas9 in a Humanized Mouse Model Ameliorates α1-antitrypsin Deficiency Phenotype. EBioMedicine 2018, 29, 104–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W.; Dutcher, S.K.; Brody, S.L. Genetics and Biology of Primary Ciliary Dyskinesia. Paediatr. Respir. Rev. 2016, 18, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boaretto, F.; Snijders, D.; Salvoro, C.; Spalletta, A.; Mostacciuolo, M.L.; Collura, M.; Cazzato, S.; Girosi, D.; Silvestri, M.; Rossi, G.A.; et al. Diagnosis of Primary Ciliary Dyskinesia by a Targeted Next-Generation Sequencing Panel: Molecular and Clinical Findings in Italian Patients. J. Mol. Diagn. 2016, 18, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Hornef, N.; Olbrich, H.; Horvath, J.; Zariwala, M.A.; Fliegauf, M.; Loges, N.T.; Wildhaber, J.; Noone, P.G.; Kennedy, M.; Antonarakis, S.E.; et al. DNAH5 Mutations Are a Common Cause of Primary Ciliary Dyskinesia with Outer Dynein Arm Defects. Am. J. Respir. Crit. Care Med. 2006, 174, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Loges, N.T.; Omran, H. 14-Dynein dysfunction as a cause of primary ciliary dyskinesia and other ciliopathies. In Dyneins: Structure, Biology and Disease (Second Edition); King, S.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 316–355. ISBN 978-0-12-809470-9. [Google Scholar]
- Zietkiewicz, E.; Bukowy-Bieryłło, Z.; Voelkel, K.; Klimek, B.; Dmeńska, H.; Pogorzelski, A.; Sulikowska-Rowińska, A.; Rutkiewicz, E.; Witt, M. Mutations in radial spoke head genes and ultrastructural cilia defects in east-european cohort of primary ciliary dyskinesia patients. PLoS ONE 2012, 7, e33667. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, L.E.; Yin, W.; Rogers, T.D.; Busalacchi, K.B.; Chua, M.; O’Neal, W.K.; Grubb, B.R. Conditional deletion of Dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 55–63. [Google Scholar] [CrossRef] [Green Version]
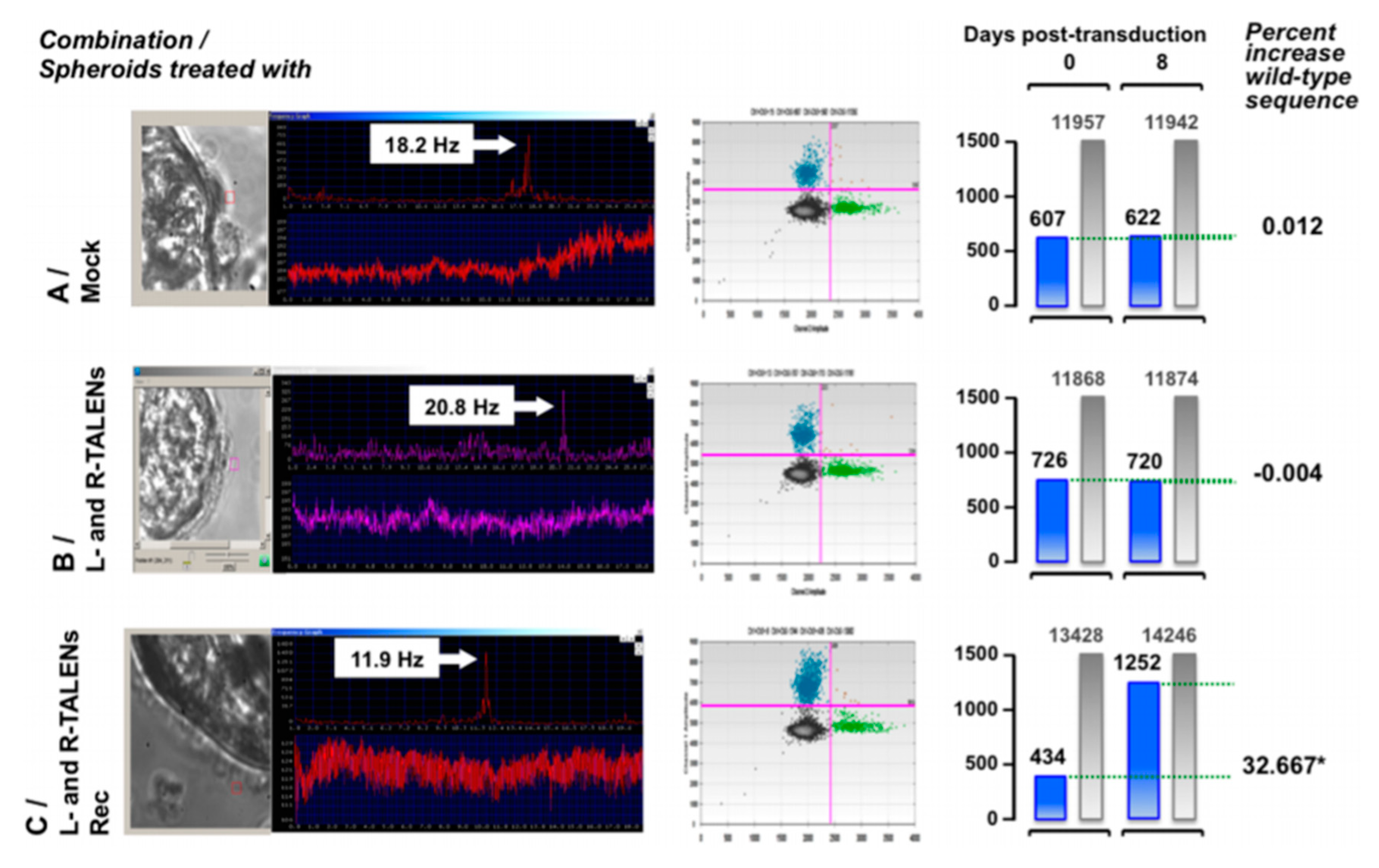
- Lai, M.; Pifferi, M.; Bush, A.; Piras, M.; Michelucci, A.; Di Cicco, M.; Del Grosso, A.; Quaranta, P.; Cursi, C.; Tantillo, E.; et al. Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. J. Med. Genet. 2016, 53, 242–249. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Vector | Genome Integration | Advantages | Disadvantages | Studies in CF, AATD, PCD |
|---|---|---|---|---|
| Lentivirus | Yes | Long-term expression | Mutagenesis potential | Yes [17], yes [18], yes [19] |
| Retrovirus | Yes | Long-term expression in dividing cells | Mutagenesis potential | Yes [20], yes [18], no |
| Adenovirus | No | Transduction is efficient in many cells | Strong antiviral immune response | Yes [21], yes [22], no |
| Adeno-associated virus | No | Non-inflammatory and non-pathogenic | Requires helper virus Small packaging capacity | Yes [21], yes [18], no |
| Herpes simplex virus | No | Large capacity | No expression when the infection is latent Tropism to neurons | No, yes [23] *, no |
| Physical Methods | Advantages | Disadvantages | Principle |
|---|---|---|---|
| Microinjection | Specific delivery Safe, Simple | Low efficiency | Uses a needle to inject the material [27] |
| Ballistic DNA | Precise delivery | Limited applications | Application of a pressurized gas to introduce nanoparticles in the cell [28] |
| Electroporation | Good efficiency Reproducible results | Low viability | Uses a high-voltage electric current to destabilize membrane polarity [29] |
| Sonoporation | Safe, Flexible | Cell damage | Ultrasounds create pores in the membrane [28] |
| Photoporation | Theoretically very efficient | Expensive Complex | Use of highly concentrated light beams that perforate the membrane [28] |
| Magnetofection | Specific delivery Used for difficult to transfect cells | Expensive Complex | Uses magnetic fields to move magnetic-sensitive particles to the cells of interest [30] |
| Hydroporation | Safe, Simple | Low efficiency Complex in large animals | Exerting osmotic pressure in the tissue environment Promotes particle movement to the interior of the cells [28] |
| Mechanical Massage | Safe, Simple | Low efficiency | Mechanical movement of the liver makes it permeable to DNA and nanoparticles [31] |
| Inorganic Particles | Advantages | Disadvantages | Principle |
|---|---|---|---|
| Calcium Phosphate | Biocompatible Biodegradable | Might crystalize when stored | Calcium is naturally absorbed by the cell [33] |
| Silica | Low toxicity Easy to store Very versatile | Interacts with serum proteins | Silica-functionalized nanoparticles are recognized and engulfed by cells [34] |
| Gold | Inert High transfection efficiency | Accumulation Long-term effects have not been studied | Its small size allows it to permeate the cell, the near infrared light absorption can be used for selective delivery [35] |
| Biocomponents | Advantages | Disadvantages | Principle |
|---|---|---|---|
| Cationic lipids | Flexible | Can become toxic at certain concentrations | Positive charges interact with the negative charged proteoglycans and glycoproteins in the membrane, helping the particles of interest enter the interior of the cells [36] |
| Lipid Nano Emulsions | Stable Very low toxicity | Less toxic than cationic lipids | |
| Solid lipid particles | Increased protection for the delivery material | Complex to produce | |
| Peptide based | Multifunctional Specific Safe | Complex to produce | Peptides can be added to lipoparticles for specific recognition or delivery [37] |
| Polyethylenimine | Widely used | Difficult to use in in vivo models | Increases osmotic pressure in the cell, creating pores in the membrane [38] |
| Chitosan | Non-toxic Mucoadhesive | Low efficiency | Increases osmotic pressure in the cell [39] |
| PLA/PLGA | Small, Phagocyted | Can induce immune reaction | Biodegradable polyesters that deliver their content by hydrolysis [40] |
| Dendrimers | Flexible, Good interaction | Toxicity | Small size allows them to interact with cell membranes, favoring DNA uptake in cells [41] |
| Polymethacrylate | Small | Poor membrane interaction | Small size allows them to reach the whole organism and deliver the content [42] |
| Clinical Trial | Vector | Administration Route | Outcome | References |
|---|---|---|---|---|
| Phase I | rAAV2-CFTR (tgAAVCG) | Nasal epithelium | Low gene transfer efficiency Safety profile | Flotte et al. (1996) |
| Phase I/II | Maxillary sinus | Dose-dependent effect Safety profile | Wagner et al. (1998) | |
| Phase II | Maxillary sinus | No significant differences Safety profile | Wagner et al. (2002) | |
| Phase I | One-dose nebulization | Safety profile | Aitken et al. (2001) | |
| Phase II | Repeated-dose nebulization | Decrease of IL-8 Improvement of FEV1 | Moss et al. (2004) | |
| Phase IIb NCT00073463 | Repeated-dose nebulization | No improvement of lung function | Moss et al. (2007) | |
| Phase I NCT00004533 | Adverse effects Minimal vector shedding PCR positive only in highest dose | Flotte et al. (2003) | ||
| Phase I/IIa NCT00789867 | pGM169/GL67A | One-dose nebulization | Safe and efficient | Alton et al. (2015) |
| Phase IIb NCT01621867 | Repeated-dose nebulization each 28 days for one year | Modest improvement in FEV1 value No adverse effects | Alton et al. (2015) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bañuls, L.; Pellicer, D.; Castillo, S.; Navarro-García, M.M.; Magallón, M.; González, C.; Dasí, F. Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far? J. Clin. Med. 2020, 9, 2577. https://doi.org/10.3390/jcm9082577
Bañuls L, Pellicer D, Castillo S, Navarro-García MM, Magallón M, González C, Dasí F. Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far? Journal of Clinical Medicine. 2020; 9(8):2577. https://doi.org/10.3390/jcm9082577
Chicago/Turabian StyleBañuls, Lucía, Daniel Pellicer, Silvia Castillo, María Mercedes Navarro-García, María Magallón, Cruz González, and Francisco Dasí. 2020. "Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far?" Journal of Clinical Medicine 9, no. 8: 2577. https://doi.org/10.3390/jcm9082577
APA StyleBañuls, L., Pellicer, D., Castillo, S., Navarro-García, M. M., Magallón, M., González, C., & Dasí, F. (2020). Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far? Journal of Clinical Medicine, 9(8), 2577. https://doi.org/10.3390/jcm9082577