Oral and Fecal Microbiota in Lynch Syndrome

 ,
,  ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Patients and Sample Collection
2.2. Microbiota Analysis
2.3. Statistical Analysis
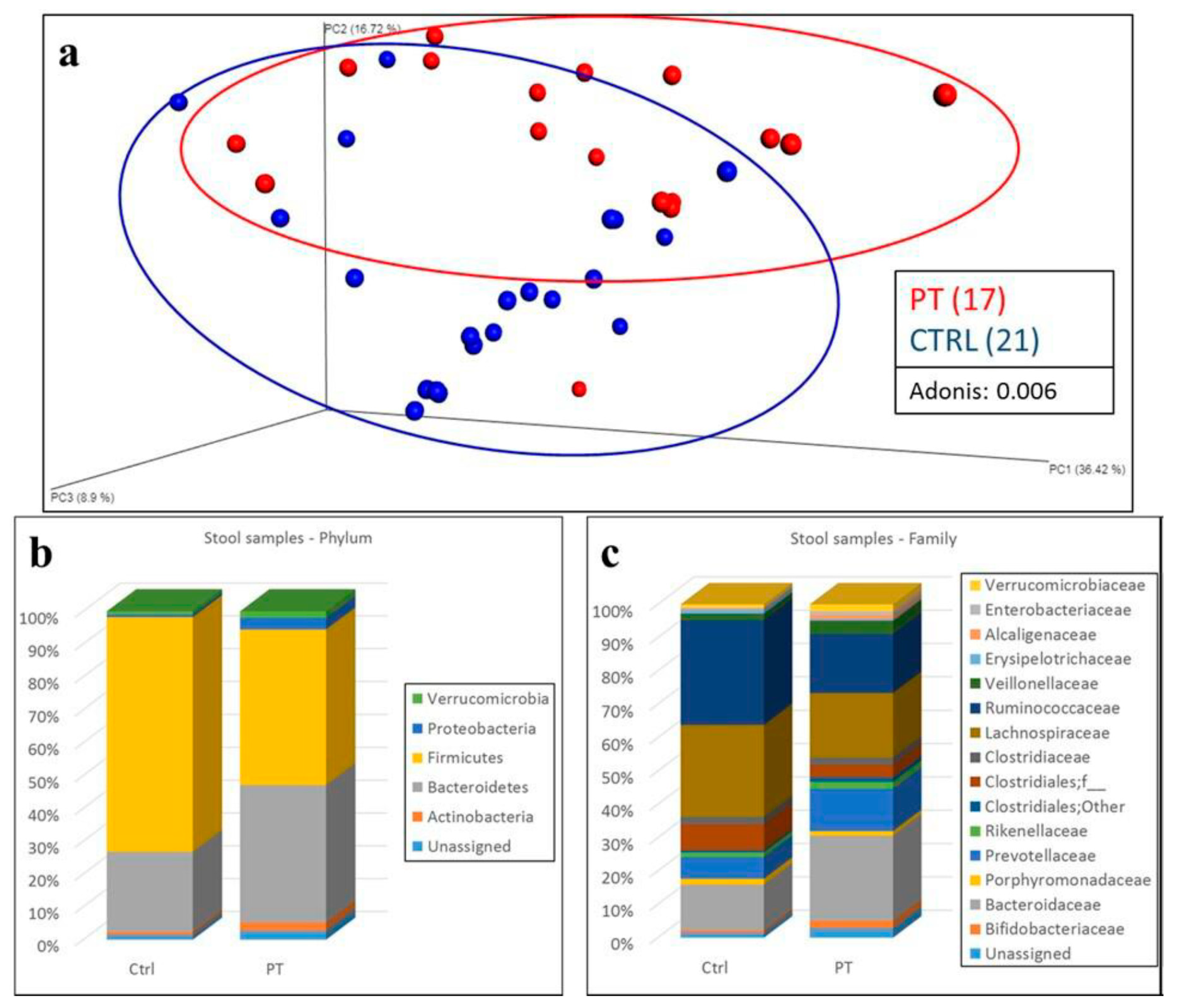
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, M.A.; Hayashi, S.; O’Shea, A.M.; Burgart, L.J.; Smyrk, T.C.; Shimizu, D.; Waring, P.M.; Ruszkiewicz, A.R.; Pollett, A.F.; Redston, M.; et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: A population-based study. Gastroenterology 2007, 133, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shia, J.; Holck, S.; Depetris, G.; Greenson, J.K.; Klimstra, D.S. Lynch syndrome-associated neoplasms: A discussion on histopathology and immunohistochemistry. Fam. Cancer 2013, 12, 241–260. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer. Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Kumar, R.; Yu, F.; Zhen, Y.H.; Li, B.; Wang, J.; Yang, Y.; Ge, H.X.; Hu, P.S.; Xiu, J. PD-1 blockade restores impaired function of ex vivo expanded CD8+ T cells and enhances apoptosis in mismatch repair deficient EpCAM + PD-L1 + cancer cells. Onco Targets Ther. 2017, 10, 3453–3465. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Shlien, A.; Campbell, B.B.; de Borja, R.; Alexandrov, L.B.; Merico, D.; Wedge, D.; Van Loo, P.; Tarpey, P.S.; Coupland, P.; Behjati, S.; et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015, 47, 257–262. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Comber, J.D.; Philip, R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther. Adv. Vaccines 2014, 2, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempski, J.; Brockmann, L.; Gagliani, N.; Huber, S. TH17 Cell and Epithelial Cell Crosstalk during Inflammatory Bowel Disease and Carcinogenesis. Front. Immunol. 2017, 8, 1373. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chiu, J.; Chen, Y.; Huang, Y.; Higashimori, A.; Fang, J.; Brim, H.; Ashktorab, H.; Ng, S.C.; Ng, S.S.M.; et al. Fecal bacteria act as novel biomarkers for noninvasive diagnosis of colorectal cancer. Clin. Cancer Res. 2017, 23, 2061–2070. [Google Scholar] [CrossRef] [Green Version]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Flynn, K.J.; Baxter, N.T.; Schloss, P.D. Metabolic and Community Synergy of Oral Bacteria in Colorectal Cancer. mSphere 2016, 11, 00102-16. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huycke, M.M. Extracellular Superoxide Production by Enterococcus faecalis Promotes Chromosomal Instability in Mammalian Cells. Gastroenterology 2007, 132, 551–561. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. Embo. Rep. 2019, 20, e47638. [Google Scholar] [CrossRef]
- Abed, J.; Emgård, J.E.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Cell Host Microbe Cell Host Microbe by Binding to Tumor-Expressed Gal-GalNAc. Cell. Host. Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Zackular, J.P.; Baxter, N.T.; Chen, G.Y.; Schloss, P.D. Manipulation of the Gut Microbiota Reveals Role in Colon Tumorigenesis. mSphere 2015, 1, e00001-15. [Google Scholar] [CrossRef] [Green Version]
- Baxter, N.T.; Zackular, J.P.; Chen, G.Y.; Schloss, P.D. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, e000145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangifesta, M.; Mancabelli, L.; Milani, C.; Gaiani, F.; de’Angelis, N.; de’Angelis, G.; van Sinderen, D.; Ventura, M.; Turroni, F. Mucosal microbiota of intestinal polyps reveals putative biomarkers of colorectal cancer. Sci. Rep. 2018, 8, 13974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [Green Version]
- Sze, M.A.; Baxter, N.T.; Ruffin, M.T., 4th; Rogers, M.A.M.; Schloss, P.D. Normalization of the microbiota in patients after treatment for colonic lesions. Microbiome 2017, 5, 150. [Google Scholar] [CrossRef]
- Sobhani, I.; Bergsten, E.; Couffin, S.; Amiot, A.; Nebbad, B.; Barau, C.; de’Angelis, N.; Rabot, S.; Canoui-Poitrine, F.; Mestivier, D.; et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc. Natl. Acad. Sci. USA 2019, 116, 24285–24295. [Google Scholar] [CrossRef]
- Yan, Y.; Drew, D.A.; Markowitz, A.; Lloyd-Price, J.; Abu-Ali, G.; Nguyen, L.H.; Tran, C.; Chung, D.C.; Gilpin, K.K.; Meixell, D.; et al. Structure of the Mucosal and Stool Microbiome in Lynch Syndrome. Cell Host Microbe 2020, 27, C585–C600. [Google Scholar] [CrossRef]
- Mori, G.; Orena, B.S.; Cultrera, I.; Barbieri, G.; Albertini, A.M.; Ranzani, G.N.; Carnevali, I.; Tibiletti, M.G.; Pasca, M.R. Gut Microbiota Analysis in Postoperative Lynch Syndrome Patients. Front. Microbiol. 2019, 10, 1746. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Kastrinos, F.; Uno, H.; Ukaegbu, C.; Alvero, C.; McFarland, A.; Yurgelun, M.B.; Kulke, M.H.; Schrag, D.; Meyerhardt, J.A.; Fuchs, C.S.; et al. Development & validation of the PREMM5 model for comprehensive risk assessment of lynch syndrome. J. Clin. Oncol. 2017, 35, 2165–2172. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Pena, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Sun, J.; Yao, F.; Lin, K.; Yuan, Y.; Chen, Y.; Han, H.; Li, Z.; Zou, J.; Jiao, X. Microbiome in Intestinal Lavage Fluid May Be A Better Indicator in Evaluating The Risk of Developing Colorectal Cancer Compared with Fecal Samples. Transl. Oncol. 2020, 13, 100772. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringel, Y.; Maharshak, N.; Ringel-Kulka, T.; Wolber, E.A.; Sartor, R.B.; Carroll, I.M. High throughput sequencing reveals distinct microbial populations within the mucosal and luminal niches in healthy individuals. Gut Microbes 2015, 6, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Wu, Y.; He, L.; Wu, L.; Wang, X.; Liu, Z. Effects of the intestinal microbial metabolite butyrate on the development of colorectal cancer. J. Cancer 2018, 9, 2510–2517. [Google Scholar] [CrossRef]
- Olsena, I.; Yamazaki, K. Can oral bacteria affect the microbiome of the gut? J. Oral. Microbiol. 2019, 11, 1586422. [Google Scholar] [CrossRef] [Green Version]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.H.; Ma, W.; Wang, D.D.; Cao, Y.; Mallick, H.; Gerbaba, T.K.; Lloyd-Price, J.; Abu-Ali, G.; Hall, A.B.; Sikavi, D.; et al. Association Between Sulfur-Metabolizing Bacterial Communities in Stool and Risk of Distal Colorectal Cancer in Men. Gastroenterology 2020, 158, 1313–1325. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gómez-Millán, J.; Queipo-Ortuño, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin, M.K., 4th; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite instability and DNA mismatch repair protein deficiency in lynch syndrome colorectal polyps. Cancer Prev. Res. 2012, 5, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Mori, T.; Ogawa, R.; Tanaka, M.; Yoshida, H.; Taniguchi, H.; Nakajima, T.; Sugano, K.; Yoshida, T.; Kato, M.; et al. Mismatch repair deficiency commonly precedes adenoma formation in Lynch Syndrome-Associated colorectal tumorigenesis. Mod. Pathol. 2017, 30, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Bläker, H. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: A pathological study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Bohaumilitzky, L.; von Knebel Doeberitz, M.; Kloor, M.; Ahadova, A. Implications of Hereditary Origin on the Immune Phenotype of Mismatch Repair-Deficient Cancers: Systematic Literature Review. J. Clin. Med. 2020, 9, 1741. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrarese, R.; Zuppardo, R.A.; Puzzono, M.; Mannucci, A.; Amato, V.; Ditonno, I.; Patricelli, M.G.; Raucci, A.R.; Clementi, M.; Elmore, U.; et al. Oral and Fecal Microbiota in Lynch Syndrome. J. Clin. Med. 2020, 9, 2735. https://doi.org/10.3390/jcm9092735
Ferrarese R, Zuppardo RA, Puzzono M, Mannucci A, Amato V, Ditonno I, Patricelli MG, Raucci AR, Clementi M, Elmore U, et al. Oral and Fecal Microbiota in Lynch Syndrome. Journal of Clinical Medicine. 2020; 9(9):2735. https://doi.org/10.3390/jcm9092735
Chicago/Turabian StyleFerrarese, Roberto, Raffaella Alessia Zuppardo, Marta Puzzono, Alessandro Mannucci, Virginia Amato, Ilaria Ditonno, Maria Grazia Patricelli, Annalisa Russo Raucci, Massimo Clementi, Ugo Elmore, and et al. 2020. "Oral and Fecal Microbiota in Lynch Syndrome" Journal of Clinical Medicine 9, no. 9: 2735. https://doi.org/10.3390/jcm9092735