Contributions of Major Cell Populations to Sjögren’s Syndrome


Abstract
1. Introduction
2. Disease Development
3. Innate Immune Cells
3.1. Dendritic Cells
3.2. Macrophages
3.3. Innate Lymphoid Cells (ILCs) and Natural Killer Cells (NK)
3.4. Salivary Gland Epithelial Cells (SGECs)
4. Adaptive Immune Cells
4.1. T cells
4.1.1. Th1 Cells
4.1.2. Th2 Cells
4.1.3. Th17 Cells
4.1.4. T Regulatory Cells (Tregs)
4.1.5. T Follicular Helper Cells (Tfh)
4.1.6. Cytotoxic T Cells/ CD8+ T Lymphocytes (CTLs)
4.2. B Cells
4.2.1. Marginal Zone B Cells
4.2.2. Memory B Cells and Plasma B Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef]
- Vivino, F.B.; Bunya, V.Y.; Massaro-Giordano, G.; Johr, C.R.; Giattino, S.L.; Schorpion, A.; Shafer, B.; Peck, A.; Sivils, K.; Rasmussen, A.; et al. Sjogren’s syndrome: An update on disease pathogenesis, clinical manifestations and treatment. Clin. Immunol. 2019, 203, 81–121. [Google Scholar] [CrossRef]
- Kassan, S.S.; Thomas, T.L.; Moutsopoulos, H.M.; Hoover, R.; Kimberly, R.P.; Budman, D.R.; Costa, J.; Decker, J.L.; Chused, T.M. Increased risk of lymphoma in sicca syndrome. Ann. Intern. Med. 1978, 89, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Risselada, A.P.; Looije, M.F.; Kruize, A.A.; Bijlsma, J.W.; van Roon, J.A. The role of ectopic germinal centers in the immunopathology of primary Sjögren’s syndrome: A systematic review. Semin. Arthritis Rheum. 2013, 42, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Kang, J.H.; Yim, Y.R.; Kim, J.E.; Lee, J.W.; Wen, L.; Park, D.J.; Kim, T.J.; Park, Y.W.; Yoon, K.C.; et al. The Significance of ectopic germinal centers in the minor salivary gland of patients with Sjögren’s Syndrome. J. Korean Med. Sci. 2016, 31, 190–195. [Google Scholar] [CrossRef]
- Reksten, T.R.; Jonsson, M.V. Sjögren’s syndrome: An update on epidemiology and current insights on pathophysiology. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hänninen, A.; Nordström, D.C. Activation of plasmacytoid dendritic cells by apoptotic particles - mechanism for the loss of immunological tolerance in Sjögren’s syndrome. Clin. Exp. Immunol. 2018, 191, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Hillen, M.R.; Ververs, F.A.; Kruize, A.A.; Van Roon, J.A. Dendritic cells, T-cells and epithelial cells: A crucial interplay in immunopathology of primary Sjögren’s syndrome. Expert Rev. Clin. Immunol. 2014, 10, 521–531. [Google Scholar] [CrossRef]
- Jonsson, R.; Nginamau, E.; Szyszko, E.; Brokstad, K.A. Role of B cells in Sjögren’s syndrome--from benign lymphoproliferation to overt malignancy. Front. Biosci. 2007, 12, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Pers, J.O.; Youinou, P. Are the B cells cast with the leading part in the Sjogren’s syndrome scenario? Oral Dis. 2014, 20, 529–537. [Google Scholar] [CrossRef]
- Singh, N.; Cohen, P.L. The T cell in Sjogren’s syndrome:F majeure, not spectateur. J. Autoimmun. 2012, 39, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kiripolsky, J.; McCabe, L.G.; Kramer, J.M. Innate immunity in Sjögren’s syndrome. Clin. Immunol. 2017, 182, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Shimizu, T.; Kawakami, A. Role of viral infections in the pathogenesis of Sjögren’s syndrome: Different characteristics of Epstein-Barr virus and HTLV-1. J. Clin. Med. 2020, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- Lessard, C.J.; Li, H.; Adrianto, I.; Ice, J.A.; Rasmussen, A.; Grundahl, K.M.; Kelly, J.A.; Dozmorov, M.G.; Miceli-Richard, C.; Bowman, S.; et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet. 2013, 45, 1284–1292. [Google Scholar] [CrossRef]
- Song, I.W.; Chen, H.C.; Lin, Y.F.; Yang, J.H.; Chang, C.C.; Chou, C.T.; Lee, M.M.; Chou, Y.C.; Chen, C.H.; Chen, Y.T.; et al. Identification of susceptibility gene associated with female primary Sjögren’s syndrome in Han Chinese by genome-wide association study. Hum. Genet. 2016, 135, 1287–1294. [Google Scholar] [CrossRef]
- Li, H.; Ice, J.A.; Lessard, C.J.; Sivils, K.L. Interferons in Sjögren’s syndrome: Genes, mechanisms, and effects. Front. Immunol. 2013, 4, 290. [Google Scholar] [CrossRef]
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type I interferon signature in Sjögren’s syndrome: Pathophysiological and clinical implications. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), 185–191. [Google Scholar]
- Brkic, Z.; Maria, N.I.; van Helden-Meeuwsen, C.G.; van de Merwe, J.P.; van Daele, P.L.; Dalm, V.A.; Wildenberg, M.E.; Beumer, W.; Drexhage, H.A.; Versnel, M.A. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann. Rheum. Dis. 2013, 72, 728–735. [Google Scholar] [CrossRef]
- Min, H.K.; Moon, S.J.; Park, K.S.; Kim, K.J. Integrated systems analysis of salivary gland transcriptomics reveals key molecular networks in Sjögren’s syndrome. Arthritis Res. Ther. 2019, 21, 294. [Google Scholar] [CrossRef]
- Nezos, A.; Gravani, F.; Tassidou, A.; Kapsogeorgou, E.K.; Voulgarelis, M.; Koutsilieris, M.; Crow, M.K.; Mavragani, C.P. Type I and II interferon signatures in Sjogren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjogren’s related lymphomagenesis. J. Autoimmun. 2015, 63, 47–58. [Google Scholar] [CrossRef]
- Maria, N.I.; Vogelsang, P.; Versnel, M.A. The clinical relevance of animal models in Sjögren’s syndrome: The interferon signature from mouse to man. Arthritis Res. Ther. 2015, 17, 172. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Rybakowska, P.D.; Dey, P.; Payerhin, K.M.; Peck, A.B.; Bagavant, H.; Deshmukh, U.S. Type I interferon receptor deficiency prevents murine Sjogren’s syndrome. J. Dent. Res. 2013, 92, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Brayer, J.; Gao, J.; Brown, V.; Killedar, S.; Yasunari, U.; Peck, A.B. A dual role for interferon-gamma in the pathogenesis of Sjogren’s syndrome-like autoimmune exocrinopathy in the nonobese diabetic mouse. Scand. J. Immunol. 2004, 60, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Meyer, O. Interferons and autoimmune disorders. Joint Bone Spine 2009, 76, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Le Page, C.; Génin, P.; Baines, M.G.; Hiscott, J. Interferon activation and innate immunity. Rev. Immunogenet. 2000, 2, 374–386. [Google Scholar]
- Mavragani, C.P.; Moutsopoulos, H.M. Sjögren’s syndrome: Old and new therapeutic targets. J. Autoimmun. 2020, 110, 102364. [Google Scholar] [CrossRef]
- Donate, A.; Voigt, A.; Nguyen, C.Q. The value of animal models to study immunopathology of primary human Sjögren’s syndrome symptoms. Expert Rev. Clin. Immunol. 2014, 10, 469–481. [Google Scholar] [CrossRef]
- Turpie, B.; Yoshimura, T.; Gulati, A.; Rios, J.D.; Dartt, D.A.; Masli, S. Sjogren’s syndrome-like ocular surface disease in thrombospondin-1 deficient mice. Am. J. Pathol. 2009, 175, 1136–1147. [Google Scholar] [CrossRef]
- Lavoie, T.N.; Lee, B.H.; Nguyen, C.Q. Current concepts: Mouse models of Sjögren’s syndrome. J. Biomed. Biotechnol. 2011, 2011, 549107. [Google Scholar] [CrossRef]
- Brayer, J.; Lowry, J.; Cha, S.; Robinson, C.P.; Yamachika, S.; Peck, A.B.; Humphreys-Beher, M.G. Alleles from chromosomes 1 and 3 of NOD mice combine to influence Sjogren’s syndrome-like autoimmune exocrinopathy. J. Rheumatol. 2000, 27, 1896–1904. [Google Scholar]
- Cha, S.; Nagashima, H.; Brown, V.B.; Peck, A.B.; Humphreys-Beher, M.G. Two NOD Idd-associated intervals contribute synergistically to the development of autoimmune exocrinopathy (Sjögren’s syndrome) on a healthy murine background. Arthritis Rheum. 2002, 46, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Roescher, N.; Lodde, B.M.; Vosters, J.L.; Tak, P.P.; Catalan, M.A.; Illei, G.G.; Chiorini, J.A. Temporal changes in salivary glands of non-obese diabetic mice as a model for Sjögren’s syndrome. Oral Dis. 2012, 18, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, P.; Jonsson, M.V.; Dalvin, S.T.; Appel, S. Role of dendritic cells in Sjögren’s syndrome. Scand. J. Immunol. 2006, 64, 219–226. [Google Scholar] [CrossRef]
- Cravens, P.D.; Lipsky, P.E. Dendritic cells, chemokine receptors and autoimmune inflammatory diseases. Immunol. Cell Biol. 2002, 80, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Choi, Y.S. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology 2005, 114, 2–10. [Google Scholar] [CrossRef]
- van Blokland, S.C.; Wierenga-Wolf, A.F.; van Helden-Meeuwsen, C.G.; Drexhage, H.A.; Hooijkaas, H.; van de Merwe, J.P.; Versnel, M.A. Professional antigen presenting cells in minor salivary glands in Sjögren’s syndrome: Potential contribution to the histopathological diagnosis? Lab. Investig. 2000, 80, 1935–1941. [Google Scholar] [CrossRef][Green Version]
- van Blokland, S.C.; van Helden-Meeuwsen, C.G.; Wierenga-Wolf, A.F.; Drexhage, H.A.; Hooijkaas, H.; van de Merwe, J.P.; Versnel, M.A. Two different types of sialoadenitis in the NOD- and MRL/lpr mouse models for Sjögren’s syndrome: A differential role for dendritic cells in the initiation of sialoadenitis? Lab. Investig. 2000, 80, 575–585. [Google Scholar] [CrossRef][Green Version]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. J. Autoimmun. 2010, 34, 400–407. [Google Scholar] [CrossRef]
- Dieu, M.C.; Vanbervliet, B.; Vicari, A.; Bridon, J.M.; Oldham, E.; Aït-Yahia, S.; Brière, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef]
- Ozaki, Y.; Amakawa, R.; Ito, T.; Iwai, H.; Tajima, K.; Uehira, K.; Kagawa, H.; Uemura, Y.; Yamashita, T.; Fukuhara, S. Alteration of peripheral blood dendritic cells in patients with primary Sjögren’s syndrome. Arthritis Rheum. 2001, 44, 419–431. [Google Scholar] [CrossRef]
- Ozaki, Y.; Ito, T.; Son, Y.; Amuro, H.; Shimamoto, K.; Sugimoto, H.; Katashiba, Y.; Ogata, M.; Miyamoto, R.; Murakami, N.; et al. Decrease of blood dendritic cells and increase of tissue-infiltrating dendritic cells are involved in the induction of Sjögren’s syndrome but not in the maintenance. Clin. Exp. Immunol. 2010, 159, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Wildenberg, M.E.; van Helden-Meeuwsen, C.G.; van de Merwe, J.P.; Moreno, C.; Drexhage, H.A.; Versnel, M.A. Lack of CCR5 on dendritic cells promotes a proinflammatory environment in submandibular glands of the NOD mouse. J. Leukoc. Biol. 2008, 83, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Vogelsang, P.; Karlsen, M.; Brun, J.G.; Jonsson, R.; Appel, S. Altered phenotype and Stat1 expression in Toll-like receptor 7/8 stimulated monocyte-derived dendritic cells from patients with primary Sjögren’s syndrome. Arthritis Res. Ther. 2014, 16, R166. [Google Scholar] [CrossRef][Green Version]
- Shi, B.; Qi, J.; Yao, G.; Feng, R.; Zhang, Z.; Wang, D.; Chen, C.; Tang, X.; Lu, L.; Chen, W.; et al. Mesenchymal stem cell transplantation ameliorates Sjögren’s syndrome via suppressing IL-12 production by dendritic cells. Stem. Cell Res. Ther. 2018, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Båve, U.; Nordmark, G.; Lövgren, T.; Rönnelid, J.; Cajander, S.; Eloranta, M.L.; Alm, G.V.; Rönnblom, L. Activation of the type I interferon system in primary Sjögren’s syndrome: A possible etiopathogenic mechanism. Arthritis Rheum. 2005, 52, 1185–1195. [Google Scholar] [CrossRef]
- Vogelsang, P.; Brun, J.G.; Oijordsbakken, G.; Skarstein, K.; Jonsson, R.; Appel, S. Levels of plasmacytoid dendritic cells and type-2 myeloid dendritic cells are reduced in peripheral blood of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1235–1238. [Google Scholar] [CrossRef] [PubMed]
- Wildenberg, M.E.; van Helden-Meeuwsen, C.G.; van de Merwe, J.P.; Drexhage, H.A.; Versnel, M.A. Systemic increase in type I interferon activity in Sjögren’s syndrome: A putative role for plasmacytoid dendritic cells. Eur. J. Immunol. 2008, 38, 2024–2033. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef]
- Hillen, M.R.; Chouri, E.; Wang, M.; Blokland, S.L.M.; Hartgring, S.A.Y.; Concepcion, A.N.; Kruize, A.A.; Burgering, B.M.T.; Rossato, M.; van Roon, J.A.G.; et al. Dysregulated miRNome of plasmacytoid dendritic cells from patients with Sjögren’s syndrome is associated with processes at the centre of their function. Rheumatology (Oxford) 2019, 58, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Hillen, M.R.; Pandit, A.; Blokland, S.L.M.; Hartgring, S.A.Y.; Bekker, C.P.J.; van der Heijden, E.H.M.; Servaas, N.H.; Rossato, M.; Kruize, A.A.; van Roon, J.A.G.; et al. Plasmacytoid DCs from patients with Sjögren’s syndrome are transcriptionally primed for enhanced pro-inflammatory cytokine production. Front. Immunol. 2019, 10, 2096. [Google Scholar] [CrossRef] [PubMed]
- Tew, J.G.; Wu, J.; Qin, D.; Helm, S.; Burton, G.F.; Szakal, A.K. Follicular dendritic cells and presentation of antigen and costimulatory signals to B cells. Immunol. Rev. 1997, 156, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.V.; Skarstein, K. Follicular dendritic cells confirm lymphoid organization in the minor salivary glands of primary Sjögren’s syndrome. J. Oral Pathol. Med. 2008, 37, 515–521. [Google Scholar] [CrossRef]
- Bombardieri, M.; Barone, F.; Humby, F.; Kelly, S.; McGurk, M.; Morgan, P.; Challacombe, S.; De Vita, S.; Valesini, G.; Spencer, J.; et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjögren’s syndrome. J. Immunol. 2007, 179, 4929–4938. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Cuello, C.; Palladinetti, P.; Tedla, N.; Di Girolamo, N.; Lloyd, A.R.; McCluskey, P.J.; Wakefield, D. Chemokine expression and leucocyte infiltration in Sjögren’s syndrome. Br. J. Rheumatol. 1998, 37, 779–783. [Google Scholar] [CrossRef]
- Hernández-Molina, G.; Michel-Peregrina, M.; Hernández-Ramírez, D.F.; Sánchez-Guerrero, J.; Llorente, L. Chemokine saliva levels in patients with primary Sjögren’s syndrome, associated Sjögren’s syndrome, pre-clinical Sjögren’s syndrome and systemic autoimmune diseases. Rheumatology (Oxford) 2011, 50, 1288–1292. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Boiu, S.; Korkolopoulou, P.; Kapsogeorgou, E.K.; Kavantzas, N.; Ziakas, P.; Patsouris, E.; Moutsopoulos, H.M. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjogren’s syndrome: Correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007, 56, 3977–3988. [Google Scholar] [CrossRef]
- Ma, W.T.; Gao, F.; Gu, K.; Chen, D.K. The role of monocytes and macrophages in autoimmune diseases: A comprehensive review. Front. Immunol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Greenwell-Wild, T.; Moutsopoulos, N.M.; Gliozzi, M.; Kapsogeorgou, E.; Rangel, Z.; Munson, P.J.; Moutsopoulos, H.M.; Wahl, S.M. Chitinases in the salivary glands and circulation of patients with Sjögren’s syndrome: Macrophage harbingers of disease severity. Arthritis Rheum. 2011, 63, 3103–3115. [Google Scholar] [CrossRef]
- Gliozzi, M.; Greenwell-Wild, T.; Jin, W.; Moutsopoulos, N.M.; Kapsogeorgou, E.; Moutsopoulos, H.M.; Wahl, S.M. A link between interferon and augmented plasmin generation in exocrine gland damage in Sjögren’s syndrome. J. Autoimmun. 2013, 40, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Yarom, N.; Dayan, D.; Buchner, A.; Vered, M. Immunoprofile of focal lymphocytic infiltration in minor salivary glands of healthy individuals. Oral Dis. 2007, 13, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Baban, B.; Liu, J.Y.; Abdelsayed, R.; Mozaffari, M.S. Reciprocal relation between GADD153 and Del-1 in regulation of salivary gland inflammation in Sjögren syndrome. Exp. Mol. Pathol. 2013, 95, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Ballantine, L.; Midgley, A.; Harris, D.; Richards, E.; Burgess, S.; Beresford, M.W. Increased soluble phagocytic receptors sMer, sTyro3 and sAxl and reduced phagocytosis in juvenile-onset systemic lupus erythematosus. Pediatr. Rheumatol. Online J. 2015, 13, 10. [Google Scholar] [CrossRef]
- Kawano, M.; Nagata, S. Efferocytosis and autoimmune disease. Int. Immunol. 2018, 30, 551–558. [Google Scholar] [CrossRef]
- Manganelli, P.; Fietta, P. Apoptosis and Sjögren syndrome. Semin. Arthritis Rheum. 2003, 33, 49–65. [Google Scholar] [CrossRef]
- Kramer, J.M. Early events in Sjögren’s syndrome pathogenesis: The importance of innate immunity in disease initiation. Cytokine 2014, 67, 92–101. [Google Scholar] [CrossRef]
- Hauk, V.; Fraccaroli, L.; Grasso, E.; Eimon, A.; Ramhorst, R.; Hubscher, O.; Pérez Leirós, C. Monocytes from Sjögren’s syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin. Exp. Immunol. 2014, 177, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Fragoulis, G.E.; Vakrakou, A.G.; Moutsopoulos, H.M. Impaired clearance of early apoptotic cells mediated by inhibitory IgG antibodies in patients with primary Sjögren’s syndrome. PLoS ONE 2014, 9, e112100. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, B.A.; Huang, Y.; Geng, X.; Dutz, J.P.; Finegood, D.T. Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes 2002, 51, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Witas, R.; Peck, A.B.; Ambrus, J.L.; Nguyen, C.Q. Sjogren’s syndrome and TAM receptors: A possible contribution to disease onset. J. Immunol. Res. 2019, 2019, 4813795. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F.; Campisi, G.; Alessandro, R.; Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren’s syndrome. Ann. Rheum. Dis. 2012, 71, 295–301. [Google Scholar] [CrossRef]
- Cortez, V.S.; Cervantes-Barragan, L.; Robinette, M.L.; Bando, J.K.; Wang, Y.; Geiger, T.L.; Gilfillan, S.; Fuchs, A.; Vivier, E.; Sun, J.C.; et al. Transforming growth factor-β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity 2016, 44, 1127–1139. [Google Scholar] [CrossRef]
- Robertson, M.J.; Ritz, J. Biology and clinical relevance of human natural killer cells. Blood 1990, 76, 2421–2438. [Google Scholar] [CrossRef]
- Seaman, W.E. Natural killer cells and natural killer T cells. Arthritis Rheum. 2000, 43, 1204–1217. [Google Scholar] [CrossRef]
- Szodoray, P.; Papp, G.; Horvath, I.F.; Barath, S.; Sipka, S.; Nakken, B.; Zeher, M. Cells with regulatory function of the innate and adaptive immune system in primary Sjögren’s syndrome. Clin. Exp. Immunol. 2009, 157, 343–349. [Google Scholar] [CrossRef]
- Sudzius, G.; Mieliauskaite, D.; Siaurys, A.; Viliene, R.; Butrimiene, I.; Characiejus, D.; Dumalakiene, I. Distribution of peripheral lymphocyte populations in primary Sjögren’s syndrome patients. J. Immunol. Res. 2015, 2015, 854706. [Google Scholar] [CrossRef]
- Struyf, N.J.; Snoeck, H.W.; Bridts, C.H.; De Clerck, L.S.; Stevens, W.J. Natural killer cell activity in Sjögren’s syndrome and systemic lupus erythematosus: Stimulation with interferons and interleukin-2 and correlation with immune complexes. Ann. Rheum. Dis. 1990, 49, 690–693. [Google Scholar] [CrossRef]
- Izumi, Y.; Ida, H.; Huang, M.; Iwanaga, N.; Tanaka, F.; Aratake, K.; Arima, K.; Tamai, M.; Kamachi, M.; Nakamura, H.; et al. Characterization of peripheral natural killer cells in primary Sjögren’s syndrome: Impaired NK cell activity and low NK cell number. J. Lab. Clin. Med. 2006, 147, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Rusakiewicz, S.; Nocturne, G.; Lazure, T.; Semeraro, M.; Flament, C.; Caillat-Zucman, S.; Sène, D.; Delahaye, N.; Vivier, E.; Chaba, K.; et al. NCR3/NKp30 contributes to pathogenesis in primary Sjogren’s syndrome. Sci. Transl. Med. 2013, 5, 195ra196. [Google Scholar] [CrossRef]
- Deshmukh, U.S.; Bagavant, H. When killers become helpers. Sci. Transl. Med. 2013, 5, 195fs129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manoussakis, M.N.; Kapsogeorgou, E.K. The role of intrinsic epithelial activation in the pathogenesis of Sjögren’s syndrome. J. Autoimmun. 2010, 35, 219–224. [Google Scholar] [CrossRef]
- Goules, A.V.; Kapsogeorgou, E.K.; Tzioufas, A.G. Insight into pathogenesis of Sjögren’s syndrome: Dissection on autoimmune infiltrates and epithelial cells. Clin. Immunol. 2017, 182, 30–40. [Google Scholar] [CrossRef]
- Tsunawaki, S.; Nakamura, S.; Ohyama, Y.; Sasaki, M.; Ikebe-Hiroki, A.; Hiraki, A.; Kadena, T.; Kawamura, E.; Kumamaru, W.; Shinohara, M.; et al. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjögren’s syndrome. J. Rheumatol. 2002, 29, 1884–1896. [Google Scholar] [PubMed]
- Jin, J.O.; Shinohara, Y.; Yu, Q. Innate immune signaling induces interleukin-7 production from salivary gland cells and accelerates the development of primary Sjögren’s syndrome in a mouse model. PLoS ONE 2013, 8, e77605. [Google Scholar] [CrossRef]
- Amft, N.; Curnow, S.J.; Scheel-Toellner, D.; Devadas, A.; Oates, J.; Crocker, J.; Hamburger, J.; Ainsworth, J.; Mathews, J.; Salmon, M.; et al. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjögren’s syndrome. Arthritis Rheum. 2001, 44, 2633–2641. [Google Scholar] [CrossRef]
- Barone, F.; Bombardieri, M.; Rosado, M.M.; Morgan, P.R.; Challacombe, S.J.; De Vita, S.; Carsetti, R.; Spencer, J.; Valesini, G.; Pitzalis, C. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren’s syndrome and MALT lymphoma: Association with reactive and malignant areas of lymphoid organization. J. Immunol. 2008, 180, 5130–5140. [Google Scholar] [CrossRef]
- Salomonsson, S.; Larsson, P.; Tengnér, P.; Mellquist, E.; Hjelmström, P.; Wahren-Herlenius, M. Expression of the B cell-attracting chemokine CXCL13 in the target organ and autoantibody production in ectopic lymphoid tissue in the chronic inflammatory disease Sjögren’s syndrome. Scand. J. Immunol. 2002, 55, 336–342. [Google Scholar] [CrossRef]
- Gong, Y.Z.; Nititham, J.; Taylor, K.; Miceli-Richard, C.; Sordet, C.; Wachsmann, D.; Bahram, S.; Georgel, P.; Criswell, L.A.; Sibilia, J.; et al. Differentiation of follicular helper T cells by salivary gland epithelial cells in primary Sjögren’s syndrome. J. Autoimmun. 2014, 51, 57–66. [Google Scholar] [CrossRef]
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Sellam, J.; Eid, P.; Lebon, P.; Pallier, C.; Lepajolec, C.; Mariette, X. Viruses induce high expression of BAFF by salivary gland epithelial cells through TLR- and type-I IFN-dependent and -independent pathways. Eur. J. Immunol. 2008, 38, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Lavie, F.; Miceli-Richard, C.; Quillard, J.; Roux, S.; Leclerc, P.; Mariette, X. Expression of BAFF (BLyS) in T cells infiltrating labial salivary glands from patients with Sjögren’s syndrome. J. Pathol. 2004, 202, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Aqrawi, L.A.; Kvarnström, M.; Brokstad, K.A.; Jonsson, R.; Skarstein, K.; Wahren-Herlenius, M. Ductal epithelial expression of Ro52 correlates with inflammation in salivary glands of patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2014, 177, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Katsiougiannis, S.; Tenta, R.; Skopouli, F.N. Autoimmune epithelitis (Sjögren’s syndrome); the impact of metabolic status of glandular epithelial cells on auto-immunogenicity. J. Autoimmun. 2019, 104, 102335. [Google Scholar] [CrossRef]
- Kawakami, A.; Nakashima, K.; Tamai, M.; Nakamura, H.; Iwanaga, N.; Fujikawa, K.; Aramaki, T.; Arima, K.; Iwamoto, N.; Ichinose, K.; et al. Toll-like receptor in salivary glands from patients with Sjögren’s syndrome: Functional analysis by human salivary gland cell line. J. Rheumatol. 2007, 34, 1019–1026. [Google Scholar]
- Spachidou, M.P.; Bourazopoulou, E.; Maratheftis, C.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Tzioufas, A.G.; Manoussakis, M.N. Expression of functional Toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakamura, H.; Takatani, A.; Umeda, M.; Horai, Y.; Kurushima, S.; Michitsuji, T.; Nakashima, Y.; Kawakami, A. Activation of Toll-like receptor 7 signaling in labial salivary glands of primary Sjögren’s syndrome patients. Clin. Exp. Immunol. 2019, 196, 39–51. [Google Scholar] [CrossRef]
- Deshmukh, U.S.; Nandula, S.R.; Thimmalapura, P.R.; Scindia, Y.M.; Bagavant, H. Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J. Oral Pathol. Med. 2009, 38, 42–47. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary epithelial cells from Sjogren’s syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J. Autoimmun. 2010, 35, 212–218. [Google Scholar] [CrossRef]
- Nakamura, H.; Horai, Y.; Suzuki, T.; Okada, A.; Ichinose, K.; Yamasaki, S.; Koji, T.; Kawakami, A. TLR3-mediated apoptosis and activation of phosphorylated Akt in the salivary gland epithelial cells of primary Sjogren’s syndrome patients. Rheumatol. Int. 2013, 33, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yang, Y.; Jin, Y.; Zhao, R.; Dong, C.; Zheng, W.; Zhang, T.; Li, J.; Gu, Z. B7-H3 participates in human salivary gland epithelial cells apoptosis through NF-κB pathway in primary Sjögren’s syndrome. J. Transl. Med. 2019, 17, 268. [Google Scholar] [CrossRef] [PubMed]
- Vakrakou, A.G.; Polyzos, A.; Kapsogeorgou, E.K.; Thanos, D.; Manoussakis, M.N. Impaired anti-inflammatory activity of PPARγ in the salivary epithelia of Sjögren’s syndrome patients imposed by intrinsic NF-κB activation. J. Autoimmun. 2018, 86, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Wahren-Herlenius, M. Update on the immunobiology of Sjögren’s syndrome. Curr. Opin. Rheumatol. 2015, 27, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Rosloniec, E.F.; Latham, K.; Guedez, Y.B. Paradoxical roles of IFN-gamma in models of Th1-mediated autoimmunity. Arthritis Res. 2002, 4, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.; Fletcher, C.; Ng, L.G.; Groom, J.; Wheway, J.; Laâbi, Y.; Xin, X.; Schneider, P.; Tschopp, J.; Mackay, C.R.; et al. TNF deficiency fails to protect BAFF transgenic mice against autoimmunity and reveals a predisposition to B cell lymphoma. J. Immunol. 2004, 172, 812–822. [Google Scholar] [CrossRef]
- Karabiyik, A.; Peck, A.B.; Nguyen, C.Q. The important role of T cells and receptor expression in Sjogren’s syndrome. Scand. J. Immunol. 2013, 78, 157–166. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Sharma, A.; Lee, B.H.; She, J.X.; McIndoe, R.A.; Peck, A.B. Differential gene expression in the salivary gland during development and onset of xerostomia in Sjogren’s syndrome-like disease of the C57BL/6.NOD-Aec1Aec2 mouse. Arthritis Res. Ther. 2009, 11, R56. [Google Scholar] [CrossRef]
- Zheng, Y.; Yang, W.; Aldape, K.; He, J.; Lu, Z. Epidermal growth factor (EGF)-enhanced vascular cell adhesion molecule-1 (VCAM-1) expression promotes macrophage and glioblastoma cell interaction and tumor cell invasion. J. Biol. Chem. 2013, 288, 31488–31495. [Google Scholar] [CrossRef]
- Walsh, S.V.; Hopkins, A.M.; Nusrat, A. Modulation of tight junction structure and function by cytokines. Adv. Drug Deliv. Rev. 2000, 41, 303–313. [Google Scholar] [CrossRef]
- Ewert, P.; Aguilera, S.; Alliende, C.; Kwon, Y.J.; Albornoz, A.; Molina, C.; Urzúa, U.; Quest, A.F.; Olea, N.; Pérez, P.; et al. Disruption of tight junction structure in salivary glands from Sjögren’s syndrome patients is linked to proinflammatory cytokine exposure. Arthritis Rheum. 2010, 62, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Abu-Helu, R.F.; Dimitriou, I.D.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Manoussakis, M.N. Induction of salivary gland epithelial cell injury in Sjogren’s syndrome: In vitro assessment of T cell-derived cytokines and Fas protein expression. J. Autoimmun. 2001, 17, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Chaly, Y.; Barr, J.Y.; Sullivan, D.A.; Thomas, H.E.; Brodnicki, T.C.; Lieberman, S.M. Type I interferon signaling is required for dacryoadenitis in the nonobese diabetic mouse model of Sjögren syndrome. Int. J. Mol. Sci. 2018, 19, 3259. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Kawai, T.; Cha, S.; Yu, Q. Interleukin-7 enhances the Th1 response to promote the development of Sjögren’s syndrome-like autoimmune exocrinopathy in mice. Arthritis Rheum. 2013, 65, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Iwai, Y.; Oh-Hora, M.; Yamamoto, M.; Morio, T.; Aoki, K.; Ohya, K.; Jetten, A.M.; Akira, S.; Muta, T.; et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 2010, 464, 1381–1385. [Google Scholar] [CrossRef]
- Okuma, A.; Hoshino, K.; Ohba, T.; Fukushi, S.; Aiba, S.; Akira, S.; Ono, M.; Kaisho, T.; Muta, T. Enhanced apoptosis by disruption of the STAT3-IκB-ζ signaling pathway in epithelial cells induces Sjögren’s syndrome-like autoimmune disease. Immunity 2013, 38, 450–460. [Google Scholar] [CrossRef]
- Forbes, L.R.; Milner, J.; Haddad, E. Signal transducer and activator of transcription 3: A year in review. Curr. Opin. Hematol. 2016, 23, 23–27. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Zhou, L.; Littman, D.R. Transcriptional regulation of Th17 cell differentiation. Semin. Immunol. 2007, 19, 409–417. [Google Scholar] [CrossRef]
- Sakai, A.; Sugawara, Y.; Kuroishi, T.; Sasano, T.; Sugawara, S. Identification of IL-18 and Th17 cells in salivary glands of patients with Sjogren’s syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J. Immunol. 2008, 181, 2898–2906. [Google Scholar] [CrossRef]
- Bombardieri, M.; Barone, F.; Pittoni, V.; Alessandri, C.; Conigliaro, P.; Blades, M.C.; Priori, R.; McInnes, I.B.; Valesini, G.; Pitzalis, C. Increased circulating levels and salivary gland expression of interleukin-18 in patients with Sjogren’s syndrome: Relationship with autoantibody production and lymphoid organization of the periductal inflammatory infiltrate. Arthritis Res. Ther. 2004, 6, R447–R456. [Google Scholar] [CrossRef] [PubMed]
- Delaleu, N.; Immervoll, H.; Cornelius, J.; Jonsson, R. Biomarker profiles in serum and saliva of experimental Sjogren’s syndrome: Associations with specific autoimmune manifestations. Arthritis Res. Ther. 2008, 10, R22. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.; Maggi, E.; Romagnani, S. Human Th1 and Th2 cells: Functional properties, mechanisms of regulation, and role in disease. Lab Investig. 1994, 70, 299–306. [Google Scholar] [PubMed]
- León, B.; Ballesteros-Tato, A.; Lund, F.E. Dendritic cells and B cells: Unexpected partners in Th2 development. J. Immunol. 2014, 193, 1531–1537. [Google Scholar] [CrossRef]
- Hartenstein, B.; Teurich, S.; Hess, J.; Schenkel, J.; Schorpp-Kistner, M.; Angel, P. Th2 cell-specific cytokine expression and allergen-induced airway inflammation depend on JunB. EMBO J. 2002, 21, 6321–6329. [Google Scholar] [CrossRef]
- Zheng, W.; Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997, 89, 587–596. [Google Scholar] [CrossRef]
- Brayer, J.B.; Cha, S.; Nagashima, H.; Yasunari, U.; Lindberg, A.; Diggs, S.; Martinez, J.; Goa, J.; Humphreys-Beher, M.G.; Peck, A.B. IL-4-dependent effector phase in autoimmune exocrinopathy as defined by the NOD.IL-4-gene knockout mouse model of Sjögren’s syndrome. Scand. J. Immunol. 2001, 54, 133–140. [Google Scholar] [CrossRef]
- Gao, J.; Killedar, S.; Cornelius, J.G.; Nguyen, C.; Cha, S.; Peck, A.B. Sjögren’s syndrome in the NOD mouse model is an interleukin-4 time-dependent, antibody isotype-specific autoimmune disease. J. Autoimmun. 2006, 26, 90–103. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Gao, J.H.; Kim, H.; Saban, D.R.; Cornelius, J.G.; Peck, A.B. IL-4-STAT6 signal transduction-dependent induction of the clinical phase of Sjogren’s syndrome-like disease of the nonobese diabetic mouse. J. Immunol. 2007, 179, 382–390. [Google Scholar] [CrossRef]
- Matsui, K.; Sano, H. T Helper 17 cells in primary Sjögren’s syndrome. J. Clin. Med. 2017, 6, 65. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 2005, 5, 521–531. [Google Scholar] [CrossRef] [PubMed]
- de Jong, E.; Suddason, T.; Lord, G.M. Translational mini-review series on Th17 cells: Development of mouse and human T helper 17 cells. Clin. Exp. Immunol. 2010, 159, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am. J. Pathol. 2009, 175, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M.; Takeda, Y.; Slominski, A.; Kang, H.S. Retinoic acid-related Orphan Receptor γ (RORγ): Connecting sterol metabolism to regulation of the immune system and autoimmune disease. Curr. Opin. Toxicol. 2018, 8, 66–80. [Google Scholar] [CrossRef]
- Schraml, B.U.; Hildner, K.; Ise, W.; Lee, W.L.; Smith, W.A.; Solomon, B.; Sahota, G.; Sim, J.; Mukasa, R.; Cemerski, S.; et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 2009, 460, 405–409. [Google Scholar] [CrossRef]
- Kang, K.Y.; Kim, H.O.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Sun, D.I.; Jhun, J.Y.; Oh, H.J.; Park, S.H.; Kim, H.Y. Impact of interleukin-21 in the pathogenesis of primary Sjögren’s syndrome: Increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res. Ther. 2011, 13, R179. [Google Scholar] [CrossRef]
- Li, L.; He, J.; Zhu, L.; Yang, Y.; Jin, Y.; Jia, R.; Liu, X.; Liu, Y.; Sun, X.; Li, Z. The clinical relevance of IL-17-producing CD4+CD161+ cell and its subpopulations in primary Sjögren’s syndrome. J. Immunol. Res. 2015, 2015, 307453. [Google Scholar] [CrossRef]
- Alunno, A.; Carubbi, F.; Bistoni, O.; Caterbi, S.; Bartoloni, E.; Bigerna, B.; Pacini, R.; Beghelli, D.; Cipriani, P.; Giacomelli, R.; et al. CD4(-)CD8(-) T-cells in primary Sjögren’s syndrome: Association with the extent of glandular involvement. J. Autoimmun. 2014, 51, 38–43. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Hu, M.H.; Li, Y.; Stewart, C.; Peck, A.B. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjogren’s syndrome: Findings in humans and mice. Arthritis Rheum. 2008, 58, 734–743. [Google Scholar] [CrossRef]
- Voigt, A.; Esfandiary, L.; Wanchoo, A.; Glenton, P.; Donate, A.; Craft, W.F.; Craft, S.L.; Nguyen, C.Q. Sexual dimorphic function of IL-17 in salivary gland dysfunction of the C57BL/6.NOD-Aec1Aec2 model of Sjogren’s syndrome. Sci. Rep. 2016, 6, 38717. [Google Scholar] [CrossRef] [PubMed]
- Wanchoo, A.; Voigt, A.; Sukumaran, S.; Stewart, C.M.; Bhattacharya, I.; Nguyen, C.Q. Single-cell analysis reveals sexually dimorphic repertoires of Interferon-gamma and IL-17A producing T cells in salivary glands of Sjogren’s syndrome mice. Sci. Rep. 2017, 7, 12512. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Rui, K.; Deng, J.; Tian, J.; Wang, X.; Wang, S.; Ko, K.H.; Jiao, Z.; Chan, V.S.; Lau, C.S.; et al. Th17 cells play a critical role in the development of experimental Sjogren’s syndrome. Ann. Rheum. Dis. 2015, 74, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Tsuboi, H.; Matsuo, N.; Asashima, H.; Hirota, T.; Kondo, Y.; Iwakura, Y.; Takahashi, S.; Matsumoto, I.; Sumida, T. A crucial role of RORγt in the development of spontaneous Sialadenitis-like Sjögren’s syndrome. J. Immunol. 2015, 194, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Carcamo, W.C.; Chiorini, J.A.; Peck, A.B.; Nguyen, C.Q. Gene therapy using IL-27 ameliorates Sjögren’s syndrome-like autoimmune exocrinopathy. Arthritis Res. Ther. 2012, 14, R172. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Wakamatsu, E.; Tsuboi, H.; Nakamura, Y.; Hayashi, T.; Matsui, M.; Goto, D.; Ito, S.; Matsumoto, I.; Sumida, T. Pathogenic role of immune response to M3 muscarinic acetylcholine receptor in Sjogren’s syndrome-like sialoadenitis. J. Autoimmun. 2010, 35, 383–389. [Google Scholar] [CrossRef]
- Iizuka, M.; Tsuboi, H.; Matsuo, N.; Kondo, Y.; Asashima, H.; Matsui, M.; Matsumoto, I.; Sumida, T. The crucial roles of IFN-γ in the development of M3 muscarinic acetylcholine receptor induced Sjögren’s syndrome-like sialadenitis. Mod. Rheumatol. 2013, 23, 614–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, C.; Chen, H.; Li, Y.; Jin, Y.; Qi, H. Analysis of Th17-associated cytokines and clinical correlations in patients with dry eye disease. PLoS ONE 2017, 12, e0173301. [Google Scholar] [CrossRef]
- De Paiva, C.S.; Hwang, C.S.; Pitcher, J.D.; Pangelinan, S.B.; Rahimy, E.; Chen, W.; Yoon, K.C.; Farley, W.J.; Niederkorn, J.Y.; Stern, M.E.; et al. Age-related T-cell cytokine profile parallels corneal disease severity in Sjogren’s syndrome-like keratoconjunctivitis sicca in CD25KO mice. Rheumatology (Oxford) 2010, 49, 246–258. [Google Scholar] [CrossRef]
- Grosskreutz, C.L.; Hockey, H.U.; Serra, D.; Dryja, T.P. Dry eye signs and symptoms persist during systemic neutralization of IL-1β by canakinumab or IL-17A by secukinumab. Cornea 2015, 34, 1551–1556. [Google Scholar] [CrossRef]
- Ramsdell, F.; Ziegler, S.F. FOXP3 and scurfy: How it all began. Nat. Rev. Immunol. 2014, 14, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sarigul, M.; Yazisiz, V.; Bassorgun, C.I.; Ulker, M.; Avci, A.B.; Erbasan, F.; Gelen, T.; Gorczynski, R.M.; Terzioglu, E. The numbers of Foxp3 + Treg cells are positively correlated with higher grade of infiltration at the salivary glands in primary Sjogren’s syndrome. Lupus 2010, 19, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, N.M.; Moutsopoulos, H.M. Foxp3+ T-regulatory cells in Sjogren’s syndrome: Correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. Am. J. Pathol. 2008, 173, 1389–1396. [Google Scholar] [CrossRef]
- Li, X.; Qian, L.; Wang, G.; Zhang, H.; Wang, X.; Chen, K.; Zhai, Z.; Li, Q.; Wang, Y.; Harris, D.C. T regulatory cells are markedly diminished in diseased salivary glands of patients with primary Sjögren’s syndrome. J. Rheumatol. 2007, 34, 2438–2445. [Google Scholar] [PubMed]
- Liu, M.F.; Lin, L.H.; Weng, C.T.; Weng, M.Y. Decreased CD4+CD25+bright T cells in peripheral blood of patients with primary Sjogren’s syndrome. Lupus 2008, 17, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Banica, L.; Besliu, A.; Pistol, G.; Stavaru, C.; Ionescu, R.; Forsea, A.M.; Tanaseanu, C.; Dumitrache, S.; Otelea, D.; Tamsulea, I.; et al. Quantification and molecular characterization of regulatory T cells in connective tissue diseases. Autoimmunity 2009, 42, 41–49. [Google Scholar] [CrossRef]
- Alunno, A.; Petrillo, M.G.; Nocentini, G.; Bistoni, O.; Bartoloni, E.; Caterbi, S.; Bianchini, R.; Baldini, C.; Nicoletti, I.; Riccardi, C.; et al. Characterization of a new regulatory CD4+ T cell subset in primary Sjögren’s syndrome. Rheumatology (Oxford) 2013, 52, 1387–1396. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Lavie, F.; Abbed, K.; Gasnault, J.; Le Nevot, E.; Delfraissy, J.F.; Taoufik, Y.; Mariette, X. CD4 CD25high regulatory T cells are not impaired in patients with primary Sjögren’s syndrome. J. Autoimmun. 2005, 24, 235–242. [Google Scholar] [CrossRef]
- Miyara, M.; Amoura, Z.; Parizot, C.; Badoual, C.; Dorgham, K.; Trad, S.; Nochy, D.; Debré, P.; Piette, J.C.; Gorochov, G. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J. Immunol. 2005, 175, 8392–8400. [Google Scholar] [CrossRef]
- Furuzawa-Carballeda, J.; Hernández-Molina, G.; Lima, G.; Rivera-Vicencio, Y.; Férez-Blando, K.; Llorente, L. Peripheral regulatory cells immunophenotyping in primary Sjögren’s syndrome: A cross-sectional study. Arthritis Res. Ther. 2013, 15, R68. [Google Scholar] [CrossRef]
- Coursey, T.G.; Bian, F.; Zaheer, M.; Pflugfelder, S.C.; Volpe, E.A.; de Paiva, C.S. Age-related spontaneous lacrimal keratoconjunctivitis is accompanied by dysfunctional T regulatory cells. Mucosal Immunol. 2017, 10, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Maria, N.I.; van Helden-Meeuwsen, C.G.; Brkic, Z.; Paulissen, S.M.; Steenwijk, E.C.; Dalm, V.A.; van Daele, P.L.; Martin van Hagen, P.; Kroese, F.G.; van Roon, J.A.; et al. Association of increased Treg cell levels with elevated Indoleamine 2,3-Dioxygenase activity and an imbalanced Kynurenine pathway in interferon-positive primary Sjögren’s syndrome. Arthritis Rheumatol. 2016, 68, 1688–1699. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ma, J.F.; Chang, C.; Xu, T.; Gao, C.Y.; Gershwin, M.E.; Lian, Z.X. Immunobiology of T Cells in Sjögren’s syndrome. Clin. Rev. Allergy Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Chung, Y.; Martinez, G.J.; Yang, X.O.; Tanaka, S.; Matskevitch, T.D.; Wang, Y.H.; Dong, C. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Kroese, F.G.; Meiners, P.M.; Corneth, O.B.; Huitema, M.G.; Haacke, E.A.; van der Vegt, B.; Arends, S.; Vissink, A.; Bootsma, H.; et al. B cell depletion therapy normalizes circulating follicular Th cells in primary Sjögren syndrome. J. Rheumatol. 2017, 44, 49–58. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular helper T cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- Szabo, K.; Papp, G.; Dezso, B.; Zeher, M. The histopathology of labial salivary glands in primary Sjögren’s syndrome: Focusing on follicular helper T cells in the inflammatory infiltrates. Mediat. Inflamm. 2014, 2014, 631787. [Google Scholar] [CrossRef]
- Moens, L.; Tangye, S.G. Cytokine-Mediated Regulation of Plasma Cell Generation: IL-21 Takes Center Stage. Front. Immunol. 2014, 5, 65. [Google Scholar] [CrossRef]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef]
- Deenick, E.K.; Ma, C.S. The regulation and role of T follicular helper cells in immunity. Immunology 2011, 134, 361–367. [Google Scholar] [CrossRef]
- Santamaria, P. Effector lymphocytes in autoimmunity. Curr. Opin. Immunol. 2001, 13, 663–669. [Google Scholar] [CrossRef]
- Gao, C.Y.; Yao, Y.; Li, L.; Yang, S.H.; Chu, H.; Tsuneyama, K.; Li, X.M.; Gershwin, M.E.; Lian, Z.X. Tissue-resident memory CD8+ T cells acting as mediators of salivary gland damage in a murine model of Sjögren’s syndrome. Arthritis Rheumatol. 2019, 71, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Mingueneau, M.; Boudaoud, S.; Haskett, S.; Reynolds, T.L.; Nocturne, G.; Norton, E.; Zhang, X.; Constant, M.; Park, D.; Wang, W.; et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjögren’s signature correlating with disease activity and glandular inflammation. J. Allergy Clin. Immunol. 2016, 137, 1809–1821.e1812. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, S.; Suzuki, K.; Nishikawa, A.; Kassai, Y.; Takiguchi, M.; Kurisu, R.; Okuzono, Y.; Miyazaki, T.; Takeshita, M.; Yoshimoto, K.; et al. Multiomic disease signatures converge to cytotoxic CD8 T cells in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 1458–1466. [Google Scholar] [CrossRef]
- Caldeira-Dantas, S.; Furmanak, T.; Smith, C.; Quinn, M.; Teos, L.Y.; Ertel, A.; Kurup, D.; Tandon, M.; Alevizos, I.; Snyder, C.M. The chemokine receptor CXCR3 promotes CD8. J. Immunol. 2018, 200, 1133–1145. [Google Scholar] [CrossRef]
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjögren syndrome. Immunol. Cell Biol. 2017, 95, 684–694. [Google Scholar] [CrossRef]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef]
- Martin, F.; Kearney, J.F. Marginal-zone B cells. Nat. Rev. Immunol. 2002, 2, 323–335. [Google Scholar] [CrossRef]
- Grasseau, A.; Boudigou, M.; Le Pottier, L.; Chriti, N.; Cornec, D.; Pers, J.O.; Renaudineau, Y.; Hillion, S. Innate B cells: The archetype of protective immune cells. Clin. Rev. Allergy Immunol. 2020, 58, 92–106. [Google Scholar] [CrossRef]
- Weill, J.C.; Weller, S.; Reynaud, C.A. Human marginal zone B cells. Annu. Rev. Immunol. 2009, 27, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Martin, F.; Forbush, K.A.; Perlmutter, R.M.; Kearney, J.F. Evidence for selection of a population of multi-reactive B cells into the splenic marginal zone. Int. Immunol. 1997, 9, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Peeva, E.; Michael, D.; Cleary, J.; Rice, J.; Chen, X.; Diamond, B. Prolactin modulates the naive B cell repertoire. J. Clin. Investig. 2003, 111, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.M.; Michael, D.J.; Diamond, B. Cutting edge: Expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J. Immunol. 2001, 167, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Ni, D.; Weigert, M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J. Exp. Med. 2002, 196, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Voulgarelis, M.; Ziakas, P.D.; Papageorgiou, A.; Baimpa, E.; Tzioufas, A.G.; Moutsopoulos, H.M. Prognosis and outcome of non-Hodgkin lymphoma in primary Sjögren syndrome. Medicine (Baltimore) 2012, 91, 1–9. [Google Scholar] [CrossRef]
- Daridon, C.; Pers, J.O.; Devauchelle, V.; Martins-Carvalho, C.; Hutin, P.; Pennec, Y.L.; Saraux, A.; Youinou, P. Identification of transitional type II B cells in the salivary glands of patients with Sjögren’s syndrome. Arthritis Rheum. 2006, 54, 2280–2288. [Google Scholar] [CrossRef]
- Groom, J.; Kalled, S.L.; Cutler, A.H.; Olson, C.; Woodcock, S.A.; Schneider, P.; Tschopp, J.; Cachero, T.G.; Batten, M.; Wheway, J.; et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren’s syndrome. J. Clin. Investig. 2002, 109, 59–68. [Google Scholar] [CrossRef]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef]
- Fletcher, C.A.; Sutherland, A.P.; Groom, J.R.; Batten, M.L.; Ng, L.G.; Gommerman, J.; Mackay, F. Development of nephritis but not sialadenitis in autoimmune-prone BAFF transgenic mice lacking marginal zone B cells. Eur. J. Immunol. 2006, 36, 2504–2514. [Google Scholar] [CrossRef]
- Shen, L.; Gao, C.; Suresh, L.; Xian, Z.; Song, N.; Chaves, L.D.; Yu, M.; Ambrus, J.L., Jr. Central role for marginal zone B cells in an animal model of Sjogren’s syndrome. Clin. Immunol. 2016, 168, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, M.; Romero, X.; Sintes, J.; Terhorst, C.; Engel, P. Targeting of Ly9 (CD229) disrupts marginal zone and B1 B cell homeostasis and antibody responses. J. Immunol. 2016, 196, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Puñet-Ortiz, J.; Sáez Moya, M.; Cuenca, M.; Caleiras, E.; Lazaro, A.; Engel, P. Ly9 (CD229) antibody targeting depletes marginal zone and germinal center B cells in lymphoid tissues and reduces salivary gland inflammation in a mouse model of Sjögren’s Syndrome. Front. Immunol. 2018, 9, 2661. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Lam, K.P.; Rajewsky, K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature 2000, 407, 636–642. [Google Scholar] [CrossRef]
- Schittek, B.; Rajewsky, K. Maintenance of B-cell memory by long-lived cells generated from proliferating precursors. Nature 1990, 346, 749–751. [Google Scholar] [CrossRef]
- Giesecke, C.; Frölich, D.; Reiter, K.; Mei, H.E.; Wirries, I.; Kuhly, R.; Killig, M.; Glatzer, T.; Stölzel, K.; Perka, C.; et al. Tissue distribution and dependence of responsiveness of human antigen-specific memory B cells. J. Immunol. 2014, 192, 3091–3100. [Google Scholar] [CrossRef]
- Steiniger, B.; Timphus, E.M.; Jacob, R.; Barth, P.J. CD27+ B cells in human lymphatic organs: Re-evaluating the splenic marginal zone. Immunology 2005, 116, 429–442. [Google Scholar] [CrossRef]
- Manzo, A.; Vitolo, B.; Humby, F.; Caporali, R.; Jarrossay, D.; Dell’accio, F.; Ciardelli, L.; Uguccioni, M.; Montecucco, C.; Pitzalis, C. Mature antigen-experienced T helper cells synthesize and secrete the B cell chemoattractant CXCL13 in the inflammatory environment of the rheumatoid joint. Arthritis Rheum. 2008, 58, 3377–3387. [Google Scholar] [CrossRef]
- Rupprecht, T.A.; Plate, A.; Adam, M.; Wick, M.; Kastenbauer, S.; Schmidt, C.; Klein, M.; Pfister, H.W.; Koedel, U. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J. Neuroinflamm. 2009, 6, 42. [Google Scholar] [CrossRef]
- Sáez de Guinoa, J.; Barrio, L.; Mellado, M.; Carrasco, Y.R. CXCL13/CXCR5 signaling enhances BCR-triggered B-cell activation by shaping cell dynamics. Blood 2011, 118, 1560–1569. [Google Scholar] [CrossRef]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, antibodies, and more. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Fazilleau, N.; McHeyzer-Williams, L.J.; Rosen, H.; McHeyzer-Williams, M.G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009, 10, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Moisini, I.; Davidson, A. BAFF: A local and systemic target in autoimmune diseases. Clin. Exp. Immunol. 2009, 158, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ittah, M.; Miceli-Richard, C.; Eric Gottenberg, J.; Lavie, F.; Lazure, T.; Ba, N.; Sellam, J.; Lepajolec, C.; Mariette, X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjögren’s syndrome. Arthritis Res. Ther. 2006, 8, R51. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Ramos-Casals, M.; Nardi, N.; Cervera, R.; Yagüe, J.; Ingelmo, M.; Font, J. Circulating monoclonal immunoglobulins in Sjögren syndrome: Prevalence and clinical significance in 237 patients. Medicine (Baltimore) 2005, 84, 90–97. [Google Scholar] [CrossRef]
- Szyszko, E.A.; Brokstad, K.A.; Oijordsbakken, G.; Jonsson, M.V.; Jonsson, R.; Skarstein, K. Salivary glands of primary Sjogren’s syndrome patients express factors vital for plasma cell survival. Arthritis Res. Ther. 2011, 13, R2. [Google Scholar] [CrossRef]
- Tengnér, P.; Halse, A.K.; Haga, H.J.; Jonsson, R.; Wahren-Herlenius, M. Detection of anti-Ro/SSA and anti-La/SSB autoantibody-producing cells in salivary glands from patients with Sjögren’s syndrome. Arthritis Rheum. 1998, 41, 2238–2248. [Google Scholar] [CrossRef]
- Halse, A.; Harley, J.B.; Kroneld, U.; Jonsson, R. Ro/SS-A-reactive B lymphocytes in salivary glands and peripheral blood of patients with Sjögren’s syndrome. Clin. Exp. Immunol. 1999, 115, 203–207. [Google Scholar] [CrossRef]
- Aqrawi, L.A.; Skarstein, K.; Øijordsbakken, G.; Brokstad, K.A. Ro52- and Ro60-specific B cell pattern in the salivary glands of patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2013, 172, 228–237. [Google Scholar] [CrossRef]
- Aqrawi, L.A.; Brokstad, K.A.; Jakobsen, K.; Jonsson, R.; Skarstein, K. Low number of memory B cells in the salivary glands of patients with primary Sjögren’s syndrome. Autoimmunity 2012, 45, 547–555. [Google Scholar] [CrossRef]
- Szabó, K.; Papp, G.; Szántó, A.; Tarr, T.; Zeher, M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjögren’s syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2016, 183, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, present, and future of rituximab-the world’s first oncology monoclonal antibody therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Moss, B.P.; Cohen, J.A. Safety of monoclonal antibodies for the treatment of multiple sclerosis. Expert. Opin. Drug. Saf. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Blüml, S.; McKeever, K.; Ettinger, R.; Smolen, J.; Herbst, R. B-cell targeted therapeutics in clinical development. Arthritis Res. Ther. 2013, 15 (Suppl. S1), S4. [Google Scholar] [CrossRef]
- Pertovaara, M.; Antonen, J.; Hurme, M. Th2 cytokine genotypes are associated with a milder form of primary Sjogren’s syndrome. Ann. Rheum. Dis. 2006, 65, 666–670. [Google Scholar] [CrossRef]
- Fernández-Fernández, F.J. Comment on: Th17 cells and IL-17A--focus on immunopathogenesis and immunotherapeutics. Semin. Arthritis Rheum. 2014, 44, e3. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- MacLennan, I.C. Germinal centers. Annu. Rev. Immunol. 1994, 12, 117–139. [Google Scholar] [CrossRef]
- Nocturne, G.; Mariette, X. B cells in the pathogenesis of primary Sjögren syndrome. Nat. Rev. Rheumatol. 2018, 14, 133–145. [Google Scholar] [CrossRef] [PubMed]


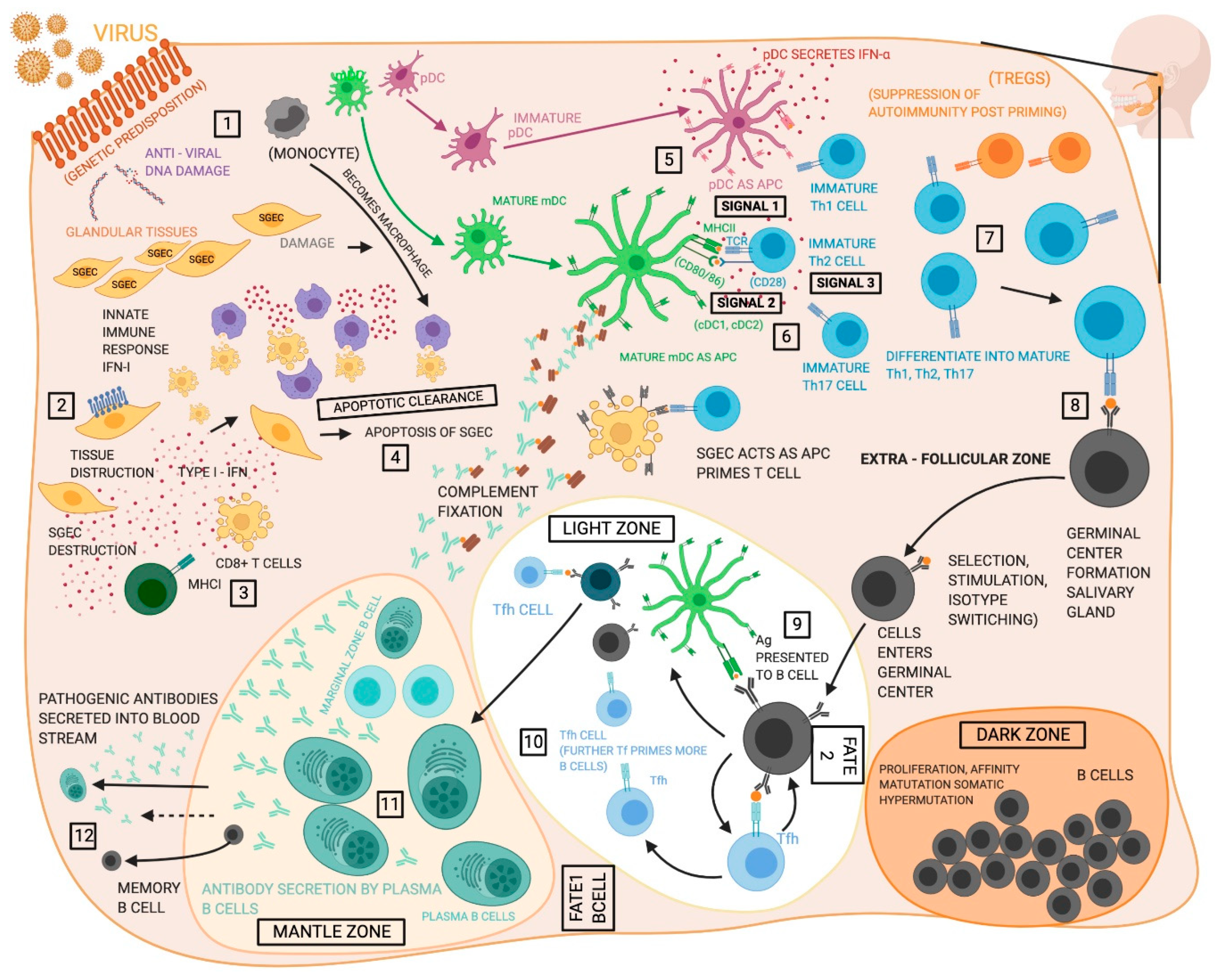{kind=link}
| Cell Type | Immunity | Function | References |
|---|---|---|---|
| Dendritic cells | Innate | • mDC are increased in pSS SGs, pSS patient mDCs have increased IL-12p40 secretion and HLA-DR expression. • pDC identified in pSS SGs, pSS patient pDCs are decreased in circulation but show increased activation. • fDC can be organized into fDC networks within functional ectopic GC in the SGs. | [41,42,44] |
| Macrophages | Innate | • Macrophage infiltration correlates with disease severity in pSS. • Infiltrating macrophages express IL-18 and proteases allowing them to contribute to inflammation and tissue destruction. • pSS monocytes and SS mouse model macrophages display impaired efferocytosis. | [38,59,61] |
| Salivary gland epithelial cells (SGECs) | Innate | • SGECs can operate as non-professional APCs and as sources of multiple inflammatory cytokines. • SGECs are sensitive to TLR induced apoptosis. | [86,101] |
| Th1 cells | Adaptive | • Play a role in the organ development of SGs. They prevent normal acinar cell proliferation and maturation. • Secrete IFN-γ that induces expression of glandular adhesion molecules allow the influx of inflammatory cells into SGs. • In-vitro exposure of acinar cells to IFN-γ causes alterations in tight junction components as observed in the SGs of patients with pSS. | [97,108,109,112] |
| Th2 cells | Adaptive | • Secrete IL-4 that prevents secretory function. • Secretion of IL-4 causes formation of IgG1 isotypic autoantibodies against M3R indicating a critical role of IgG1, IgG2a, IgG2b, IgG3, IgM, and IgA isotype switching in SS. • Stat6 gene also prevents IgG1 production against M3R and also plays a part in the isotype class switching. | [108,129,216] |
| Th17 cells | Adaptive | • They are stimulated by cytokines that play a role in the progression of the disease such as IL-22 and IL-23. • IL-22 is derived primarily from natural killer cells, is produced by Th17 cells, and it has been identified in the mSG tissue of pSS patients. • Th17 cells produce IL-17A (refer to as IL-17) and five other IL-17 members which have also been described that are termed as IL-17B, C, D, E (or IL-25), and F with conserved residues in the c-terminal region that form homodimers. • Local IL-17 protein production and mRNA levels, together with IL-6 and IL-23 mRNA, have been shown to increase with the progression of lesion severity in mSGs of pSS patients. • Conjunctival RORγT mRNA and protein expression in tears is observed to be higher in pSS as compared to non-SS patients exhibiting dry eye disease. • IL-21 expression in SGs has also been associated with hypergammaglobulinemia and patients with primary SS. | [133,134,152,155,163,217] |
| T regulatory cells (Tregs) | Adaptive | • Important for the induction and maintenance of peripheral tolerance therefore, they are key in preventing excessive immune responses in SS. • Suppressive activity towards autoreactive lymphocytes via either cell-cell contact or the release of soluble mediators that notably include IL-10 and TGF-β. • Reduction of peripheral blood Treg cells in humans that lead to exacerbated clinical symptoms of SS. Role of Tregs is uncertain because of a balance in between Tregs and Th17 cells. | [152,154,218,219] |
| T follicular helper cells (Tfh) | Adaptive | • Specialized providers of T cell help to B cells, marked increase of Bcl6 and other transcription factors that are usually upregulated in SS. • Important in the formation of GCs and primarily show presence of CD84 a cell surface marker, observed in SS. • The function of CXCR5 positive Tfh cells is directly related to the secretion of IL-21 mediating B cell maturation, proliferation, and GC formation. | [165,166] |
| Cytotoxic T cells/ CD8+ T cells (CTLs) | Adaptive | • They produce the pro-inflammatory effector cytokines TNF-α or IFN-γ. • Tissue resident memory CD8+ T cells act as mediators of SG damage in murine models of SS but the pathogenic significance of CD8+ T cells is unclear as limited studies have been performed to illuminate their role. • Tend to colocalize with salivary duct epithelial cells and acinar cells, and produce pro-inflammatory cytokines. | [172] |
| Marginal Zone B cells | Adaptive | • Stimulated by BAFF. • MZ B cells within the IL14atg model drive reduced saliva flow rate, lymphocytic infiltrations into the SGs, and formation of autoantibodies. • Possess self-reactive BCRs that cause complications of SS and the increased incidence of B cell lymphoma. | [174,179,189,192,193] |
| Memory B cells | Adaptive | • Maintain memory for SS antigens in the absence of constant antigen stimulation. • CXCL13 is the key cytokine responsible for the homing of B cells to the SGs. • CD27+ memory B cells, attract the subpopulation of peripheral CD27+ memory B cells into the inflamed glands where they reside and cause inflammation. • The primary cell type that produces antibodies, and thus are drivers of antibody-mediated immunity. • BAFF primary cytokine produced, that has a role in B cell maturation, class switching, survival, and proliferation especially in advanced disease and is produced by SGEC, DC, macrophages, activated T cells, and also B cells • Cause the formation of GCs in SGs and work antagonistically to Tfr cells. | [88,167,194,195,220] |
| Plasma B cells | Adaptive | • B cells that produce SS auto-antibodies with specific BCRs against auto-antigens after differentiation from Memory B cells or circulating peripheral B cells. | [189,203,221] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witas, R.; Gupta, S.; Nguyen, C.Q. Contributions of Major Cell Populations to Sjögren’s Syndrome. J. Clin. Med. 2020, 9, 3057. https://doi.org/10.3390/jcm9093057
Witas R, Gupta S, Nguyen CQ. Contributions of Major Cell Populations to Sjögren’s Syndrome. Journal of Clinical Medicine. 2020; 9(9):3057. https://doi.org/10.3390/jcm9093057
Chicago/Turabian StyleWitas, Richard, Shivai Gupta, and Cuong Q. Nguyen. 2020. "Contributions of Major Cell Populations to Sjögren’s Syndrome" Journal of Clinical Medicine 9, no. 9: 3057. https://doi.org/10.3390/jcm9093057
APA StyleWitas, R., Gupta, S., & Nguyen, C. Q. (2020). Contributions of Major Cell Populations to Sjögren’s Syndrome. Journal of Clinical Medicine, 9(9), 3057. https://doi.org/10.3390/jcm9093057