Abstract
Grain size is a key factor influencing the grain yield in rice. To identify the as-yet-unknown genes regulating grain size in Korean japonica rice, we developed a recombinant inbred line population (n = 162) from a cross between Odae (large-grain) and Joun (small-grain), and measured six traits including the thousand-grain weights of unhulled and hulled seeds, grain area, grain length, grain width and grain length-to-width ratio using high-throughput image analysis at the F8 and F9 generations. A genetic map was constructed using 248 kompetitive allele-specific PCR (KASP) markers that were polymorphic between the parental genotypes, and 29 QTLs affecting the six traits were identified, of which 15 were stable in both F8 and F9 generations. Notably, three QTL clusters affecting multiple traits were detected on chromosomes 6, 7 and 11. We analyzed whole-genome resequencing data of Odae and Joun, and selected candidate genes for the stable QTLs in the identified clusters that have high- or moderate-impact variations between Odae and Joun and encode proteins the families of which have been reported to be related to grain size regulation. These results will facilitate the identification of genes underlying the QTLs and promote molecular breeding of high-yielding Korean japonica rice varieties.
1. Introduction
Rice (Oryza sativa L.) is a major food crop throughout the world and a staple food of the populations in Asia, the Pacific and Latin America [1]. While the world population continues to grow, there are increasing concerns about the potential reduction in rice yield due to global warming-induced severe droughts and other extreme weather events. Therefore, the breeding of sustainable and high-yielding rice varieties is critical [2,3]. Rice yield, a complex agronomic trait, is affected by the number of panicles per unit land area, number of spikelets per panicle, percentage of filled grains and 1000-grain weight (TGW) [4,5]. Grain size, one of the most important quantitative traits directly affecting rice yield, is determined by grain length (GL), grain width (GW) and grain thickness (GT), and grain weight is largely determined by grain size. Grain size is considered a primary target trait for improving rice yield via breeding [6,7]. Recently, functional genomics analyses, based on highly accurate rice genome sequence information, led to the identification of rice genes underlying key quantitative trait loci (QTLs), including grain size 3 (GS3) [8,9], grain width 2 (GW2) [10], wide and thick grain (OsOTUB1/WTG1) [11], GS5 [12], GS2 [13], grain length 3 (GL3.1/qGL3) [14,15], GW5/qSW5 [16,17], thousand-grain weight 3 (TGW3) [18], TGW6 [19], GW6 [20], grain length and weight on chromosome 7 (GLW7) [21] and GW8 [22,23], which are closely linked to traits such as GL, GW and GT. Furthermore, functional analyses showed that genes controlling grain size act as positive or negative regulators in various signaling pathways, including the G-protein signaling, ubiquitin-proteasome and phytohormone signaling pathways, and transcriptional regulation [24].
GS3, which affects grain size via the G-protein signaling pathway, was the first QTL to be molecularly characterized. GS3 encodes a putative transmembrane protein harboring a plant-specific organ size regulation (OSR) domain, a transmembrane domain, a tumor necrosis factor receptor/nerve growth factor receptor (TNFR/NGFR) family domain and a Von Willebrand factor type C (VWFC) domain [8]. Moreover, a recent report showed that the OSR domain (a G gamma-like domain) of GS3 is sufficient for controlling both grain length and weight [9].
GW2 and OsOTUB1/WTG1 encode a RING-type E3 ubiquitin ligase and an otubain-like protease, respectively. These genes are highly associated with the ubiquitin-proteasome pathway and regulate the overall grain size in rice [10,11]. Previously, the role of different phytohormones, including brassinosteroid (BR), cytokinin, auxin and gibberellin, in grain size regulation, has been studied extensively. The following QTLs act as key players in BR signaling: GS5, which encodes a putative serine carboxypeptidase (SCP) that specifically affects grain width and filling [12]; GS2, which encodes growth-regulating factor 4 (GRF4) that regulates grain length and width [13]; GL3.1/qGL3, which encodes a putative protein phosphatase with a Kelch-like repeat domain, and affects grain length, width and weight [14,15]; and GW5/qSW5, which encodes a calmodulin-binding protein involved in controlling grain width and weight [16,17]. Two major QTLs affecting grain weight, TGW6 and TGW3 participate in the auxin signaling pathway. Indol-3-acetic acid (IAA)-glucose hydrolase (encoded by TGW6) and GSK3/SHAGGY-like kinase OsGSK5/OsSK41 (encoded by TGW3) participate in auxin signaling to control grain width and weight [18,19]. Hu et al. recently identified a gibberellin-regulated GAST family protein, GW6, which contributes to improvement in grain width and weight [20]. Transcription factor-mediated regulation represents another major pathway that contributes to grain size. For instance, the GLW7 and GW8 QTLs, which encode OsSPL13 and OsSPL16, respectively, regulate the transcription of the corresponding genes determining grain size [21,23,25]. It is important to note that GS5, GS2, GW6, GLW7 and GW8 positively regulate grain size, whereas GS3, GW2, OsOTUB1/WTG1, GL3.1/qGL3, GW5/qSW5, TGW3 and TGW6 have a negative effect on grain size by affecting cell division and cell proliferation, as well as endosperm development, and significantly influence GW, weight and thickness in rice [26,27].
Despite these remarkable findings, grain size remains a complex agronomic trait and is thought to be determined by the interplay of multiple genetic and environmental factors. Therefore, to establish precise breeding strategies for increasing rice yield, it is important to continue to explore more QTLs/genes and to investigate their molecular mechanisms and overall contributions to the regulation of grain size [7,24,26]. In Korea, because of its geographic location, farmers cultivate temperate japonica rice varieties, which are consumed as a staple food. Although many attempts have formerly been made to identify agronomically important QTLs/genes that can be used to generate high-quality rice varieties, these efforts have been challenged by the narrow range of genetic diversity among Korean japonica rice varieties [28,29]. Fortunately, recent advances in NGS technologies and molecular markers have provided an efficient way of overcoming these limitations and accelerating the identification of QTLs/genes’ underlying specific traits [30,31].
In previous studies, we performed whole-genome resequencing of 13 Korean japonica rice varieties, and identified 740,556 single-nucleotide polymorphisms (SNPs), which formed the basis of 1225 kompetitive allele-specific PCR (KASP) markers [32,33,34]. Subsequently, time- and cost-effective high-throughput analyses performed using these KASP markers greatly facilitated the identification of major QTLs and candidate genes responsible for bakanae disease resistance [34,35] and pre-harvest sprouting [36]. Despite the increasing demand for high-yielding rice varieties, previous studies mainly focused on the detection of grain yield-related QTLs using mapping populations derived from crosses between indica and japonica (or other) varieties, but rarely from crosses involving Korean temperate japonica rice varieties. Therefore, in this study, we developed a recombinant inbred line (RIL) population from a cross between two early-maturing Korean temperate japonica varieties, Odae (large-grain variety) and Joun (small-grain variety), and performed QTL mapping analysis using 248 KASP markers to identify QTLs affecting grain size. In sum, three QTL clusters for grain size-related traits were identified on chromosomes 6, 7 and 11, respectively. The results of this study broaden our understanding of the genetic basis of grain size, and provide a strong foundation for the development of molecular breeding approaches for improving grain yield in temperate japonica rice.
2. Materials and Methods
2.1. Plant Materials and Field Experiments
A total of 162 RILs derived from a cross between two Korean japonica rice varieties, Odae (large-grain variety) and Joun (small-grain variety), were cultivated and harvested over two successive years, 2019 and 2020, at which time the RILs were in F8 and F9 generations, respectively. The parental genotypes (Odae and Joun) were grown alongside the RILs in both years as a control. Twenty individual plants of each RIL were transplanted at the experimental field of the National Institute of Agricultural Sciences of the Rural Development Administration (Jeonju, Korea), and were grown during the natural rice-growing season. After harvest, the fully mature grains of each line were used for phenotypic evaluation and QTL mapping.
2.2. Phenotypic Evaluation of Grain Size-Related Traits
Approximately 300 grains in the F8 population grown in 2019 and 600 grains in the F9 populations grown in 2020 were used to measure six grain size-related traits, including UGW, HGW, GA, GL, GW and RLW. To determine the grain weight, unhulled and hulled grains (n = 100 (F8) or 200 (F9)) were weighed using an electronic scale with 0.01 g graduation, and the average values of each line were multiplied with a constant (10 (F8) or 5 (F9), respectively) to determine the weight of 1000 grains.
Next, the GA, GL and GW of the same sets of grains were measured by image analysis. A total of 100 (F8) or 200 (F9) grains were photographed together with a size standard tape of 10 mm length using a digital camera fixed to a stand, and the images were analyzed using an ImageJ macro program that allows accurate high-throughput data collection and analysis from a large number of grains [37]. The procedure used to perform image analysis is shown in Figure S1. Briefly, background objects were removed, and grain images were captured based on a color threshold. Then, the area, length and width of each grain were measured, and the RLW was calculated by dividing the length of each grain by its width. Brown rice was used for measuring almost all traits, except UGW, and all experiments were performed in triplicate. Correlation coefficients were calculated using GraphPad Prism v9 (San Diego, CA, USA).
2.3. Genetic Map Construction and QTL Mapping
DNA was extracted from 162 F8 RILs and used for genotyping with 265 KASP markers, which were polymorphic between the parental varieties and were selected from a collection of markers developed previously based on the whole-genome resequencing data of 13 Korean japonica varieties [32,33]. A genetic map was constructed on the basis of the genotype of RILs using MapDisto v1.7.0 [38] and MapChart v2.32 [39], and the distance between markers was calculated based on Kosambi’s function. QTLs related to grain size were detected by CIM using Windows QTL Cartographer v2.5 (http://brcwebportal.cos.ncsu.edu/qtlcart/WQTLCart.htm, accessed date: 16 September 2021) [40]. The LOD score threshold was determined by 1000 permutations at a probability level of 0.05. QTLs stably detected at the same map interval over two years were considered as the same QTL. Additionally, a chromosomal region harboring more than two QTLs affecting multiple traits was defined as a QTL cluster and analyzed further.
2.4. Analysis of Whole-Genome Resequencing Data of Odae and Joun
The whole-genome resequencing data of Odae and Joun generated previously [32] were analyzed according to the methods reported by Kumagai et al. [41] using Trimmomatic 0.36 [42], BWA-mem (v0.7.12) (https://sourceforge.net/projects/bio-bwa/, accessed date: 16 September 2021), Picard 2.9.0 (http://broadinstitute.github.io/picard/, accessed date: 16 September 2021), GATK (v4.1.3.0) (https://github.com/broadinstitute/gatk/, 16 September 2021) and SnpEff v4.3t [43]. Briefly, the low-quality nucleotides and adapter sequences in each read were removed using Trimmomatic. The clean reads were mapped to the IRGSP-1.0 Nipponbare reference genome sequence [44] using BWA-mem with default settings. After removing PCR duplicates with Picard 2.9.0, the variants in each sample were called using a GATK HaplotypeCaller. The variants identified in each parental variety were combined using GATK CombineGVCFs, and genotyping of parental varieties was performed using GATK GenotypeGVCFs. Hard filtering of variants was performed using GATK VariantFiltration and GATK SelectVariants with the filter “QD < 5.0, FS > 50.0, SOR > 3.0, MQ < 50.0, MQRankSum < −2.5, ReadPosRankSum < −1.0, ReadPosRankSum > 3.5”. The effect of each variant on gene function was predicted using SnpEff based on the rice genome annotation information available at RAP-DB [45]. The position, genotype and annotation of variants were extracted using SnpSift (v4.3t) [46]. Variants with high and moderate impacts were also extracted using SnpSift. The Illumina sequencing data of Odae and Joun varieties have been submitted to the Sequence Read Archive (SRA) database of the NCBI (https://www.ncbi.nlm.nih.gov/sra, accessed date: 3 November 2021) under the project number PRJNA789414.
2.5. Long-Read Sequencing of Odae and Joun
In order to produce long-read sequencing data, the genomic DNA of Odae and Joun was extracted from seedlings using the CTAB method. Sequencing was performed by DNA Link (Seoul, Korea) using the sequencing platform of Pacific Biosciences RSII with P6-C4 chemistry (PacBio, Menlo Park, CA, USA). The genomic DNA sample was sheared by gTUBEs (Covaris, MA, USA) to generate 20 kb fragments. The SMRTbell library was constructed using an SMRTbell® Express Template Preparation Kit (101-357-000) (PacBio, Menlo Park, CA, USA). Using the BluePippin (Sage Science, Beverly, MA, USA) size selection system, small fragments were removed. After a sequencing primer v4 was annealed to the SMRTbell template, DNA polymerase was bound to the complex (Sequel Binding kit 2.0). The SMRTbell library was sequenced using SMRT cells with a Sequel Sequencing Kit v2.1 (PacBio, Menlo Park, CA, USA). For Joun, high-fidelity (HiFi) sequencing technology was applied [47]. Assembly of long-read sequences was performed using the FALCON and FALCON-Unzip programs [48] for Odae and HiFiasm 0.15.1 program [49] for Joun. Among the contigs produced by long-read sequence assembly, the contigs including the QTL clusters on chromosomes 7 and 11 were selected by a BLAST search. The sequence variations between Odae and Joun in the contig sequences of these QTL clusters were detected using the MUMmer 3.23 program [50].
2.6. Selection of Candidate Genes Underlying Major QTLs
The physical interval of each QTL identified in this study was deduced according to the position of the flanking markers with a 95% confidence interval. Within each interval, genes showing sequence variations between Odae and Joun, with a high or moderate impact effects on gene function, were selected. Among these genes, those belonging to gene families that have been previously reported to be related to grain size were selected as candidate genes for the QTLs.
3. Results
3.1. Phenotypic Variation and Correlation Analysis
The RIL population (n = 162) and two parental varieties (large-grain Odae and small-grain Joun) (Figure 1a) were cultivated for two growing seasons (2019 and 2020), and the grains of F8 and F9 RILs and parental genotypes were evaluated for six traits closely associated with grain size, namely, unhulled 1000-grain weight (UGW), hulled 1000-grain weight (HGW), grain area (GA), GL, GW and ratio of grain length to width (RLW).
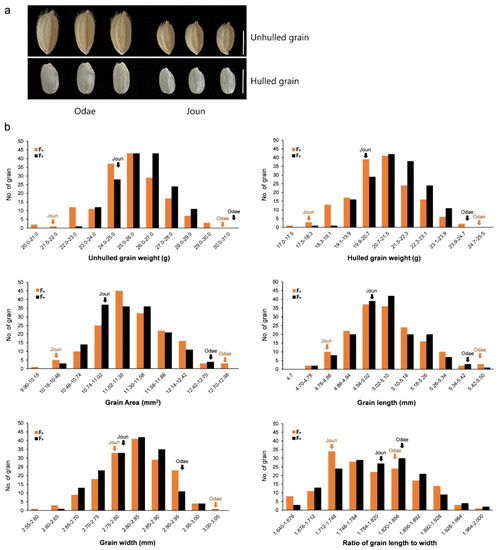
Figure 1.
Comparison of parental genotypes and frequency distributions of grain size-related traits in RIL population. (a) Grain phenotypes of parental rice varieties, Odae and Joun. Scale bar, 5 mm. (b) Frequency distributions of six grain size-related traits in F8 and F9 RIL populations (each comprising 162 lines), and two parental varieties Odae and Joun grown in 2019 and 2020. Solid arrows above the histograms indicate the frequencies of parental varieties. F8: F8 RIL population grown in 2019; F9: F9 RIL population grown in 2020; No.: number.
Phenotypic distribution analyses revealed that the data of all evaluated traits showed a normal distribution in both RIL mapping populations, with significant differences in trait values between the parental lines, indicating transgressive segregation in both RIL populations (Figure 1b). Thus, these data were suitable for QTL analysis.
Correlation coefficients of UGW, HGW, GA, GL, GW and RLW traits, calculated based on their measurements over two years, showed significant similarities between the two generations (Table 1). Two grain weight-related traits, UGW and HGW in both generations, were positively correlated with GA, GW and GL, as expected. Both UGW and HGW showed the highest positive correlation with GA (0.853 and 0.875, respectively, in F8; 0.834 and 0.806, respectively, in F9), followed by GW (0.755 and 0.767, respectively, in F8; 0.673 and 0.655, respectively, in F9) and GL (0.437 and 0.454, respectively, in F8; 0.441 and 0.421, respectively, in F9). These results suggest that grain weight, one of the most important traits determining grain size, is more strongly influenced by GW than by GL in this population. Interestingly, except GL, which showed a highly positive correlation with RLW (0.714 in F8; 0.732 in F9), negative correlations were observed between RLW and the remaining five traits, especially GW, which showed a highly negative correlation (−0.721 in F8; −0.756 in F9), even though RLW reflects GL and width.

Table 1.
Coefficients of correlation among grain size-related traits in the F8- and F9-generation populations.
3.2. Genetic Map Construction
On the basis of polymorphisms between the two parental varieties Odae and Joun, a set of 265 KASP markers were initially selected to genotype the 162 F8 RILs. While 248 of the 265 KASP markers produced reliable genotypic data, the remaining 17 markers showed poor allele discrimination, and therefore, were excluded from further analysis. Subsequently, a genetic map, based on the 248 reliable KASP markers, was successfully constructed (Figure 2). The total distance of the genetic map was 1302.8 cM, and the average distance between markers was 5.5 cM. Overall, the KASP markers were evenly distributed throughout the entire genome, although some chromosomes (e.g., chromosome 6) showed a lower marker density than others. The physical position of the 248 KASP markers is shown in Supplementary Table S1.
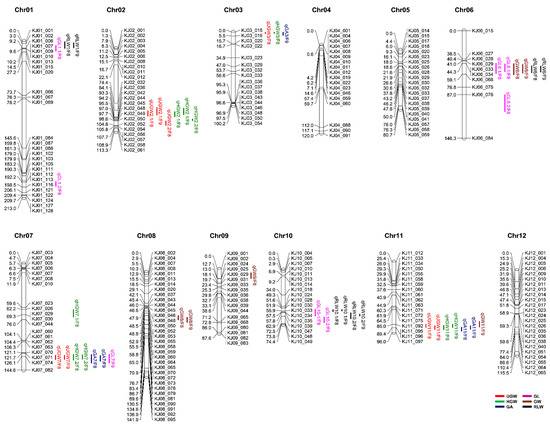
Figure 2.
Genetic map showing the chromosomal distribution of grain size-related QTLs in the RIL population (n = 162) derived from a cross between Odae and Joun. Chromosome numbers (Chr01–12) are indicated above each chromosome. Numbers on the left side of each chromosome represent the position of markers in centimorgan (cM), and IDs on the right side of each chromosome represent the KASP marker names. QTLs (vertical text) are indicated on the right side of each chromosome, at the corresponding genetic map position, and the text following a slash (/) indicates the generations of the population grown in 2019 (F8) and 2020 (F9).
3.3. Identification of Grain Size-Related QTLs
The genetic map and phenotypic data obtained from F8 and F9 populations were used for composite interval mapping (CIM) combined with a permutation test at 1000 iterations to identify QTLs closely linked to grain size. The QTLs identified were named starting with the prefix ‘q’, followed sequentially by the trait abbreviation, chromosome number and QTL serial number. To distinguish between the F8 and F9 populations on the genetic map, a slash was added to the end of the QTL name, followed by “F8” or “F9”, respectively. The QTLs were evenly distributed across all chromosomes, except chromosomes 4, 5 and 12 (Figure 2 and Table 2). In addition, QTLs with partially or fully overlapping marker intervals for each trait were considered as the same QTL.
Table 2.
Grain size-related QTLs detected in the RIL population derived from a cross between Odae and Joun.
Five QTLs related to UGW were identified on chromosomes 2, 3, 7 and 11, with two QTLs on chromosome 2 and one QTL each on chromosomes 3, 7 and 11. Moreover, three QTLs (qUGW2.1, qUGW7 and qUGW11) were detected in both F8 and F9 mapping populations, whereas qUGW2.2 and qUGW3 were identified only in the F8 population. The proportion of phenotypic variance explained by the QTL (PVE) ranged from 6% (qUGW2.1 in F9) to 24% (qUGW7 in F9), and the logarithm of the odds (LOD) score ranged from 3.3 (qUGW11 in F8) to 14.4 (qUGW7 in F9). In contrast to qUGW2.1 and qUGW2.2, which showed negative additive effects, the QTLs qUGW3, qUGW7 and qUGW11 showed positive additive effects on UGW.
Six QTLs for HGW were identified on chromosomes 2, 3, 7 and 11, of which three were major and stable QTLs found in both F8 and F9 populations. These three QTLs were as follows: qHGW2.1 (PVE values, 15% in F8 and 6% in F9; negative additive effect); qHGW7.2 (PVE values, 11% in F8 and 21% in F9; positive additive effect; highest LOD score of 12.3 in F9); and qHGW11 (PVE values, 6% in F8 and 13% in F9; positive additive effect). Notably, the three major QTLs detected for UGW overlapped with those detected for HGW. These results indicate that qHGW2.1, qHGW7.2 and qHGW11 are the major QTLs affecting grain weight, and verify the reliability of our QTL analysis.
Three QTLs, qGA3, qGA7 and qGA11 identified on chromosomes 3, 7 and 11, respectively, were associated with GA and showed positive additive effects. These QTLs were consistently detected in both F8 and F9 populations, with PVE values and LOD scores ranging from 7% to 18% and 3.7 to 8.7, respectively.
Seven QTLs associated with GL showed positive additive effects. Among these, only one QTL, qGL6.1, was identified in both F8 and F9 populations, with PVE values of 10% and 23%, respectively, while the PVE values of the other six QTLs ranged from 4% to 22%. The qGL6.1 QTL showed the highest LOD score of 10.0 in the F9 population.
Four QTLs closely associated with GW were identified on chromosomes 6, 8, 9 and 11. Among these, qGW6 and qGW8 were detected in both F8 and F9 populations, while qGW9 and qGW11 were detected only in F8 and F9, respectively. Furthermore, in contrast to qGW6, qGW8 and qGW9, which showed negative additive effects, qGW11 showed a positive additive effect on GW. The LOD scores of these QTLs ranged from 3.4 to 5.4.
Four QTLs associated with RLW were identified on chromosomes 1 (qRLW1), 6 (qRLW6) and 10 (qRLW10.1 and qRLW10.2) in both F8 and F9 mapping populations, with positive additive effects. The PVE values of these QTLs ranged from 7% to 26%, and qRLW6 showed the highest LOD score of 14.1 in the F9 population.
Overall, a total of 29 putative QTLs associated with the UGW, HGW, GA, GL, GW and RLW traits were identified in this study. Among these, 15 were detected in both the F8 and F9 populations, whereas nine and five minor QTLs were detected in only F8 and F9, respectively. Moreover, several QTLs linked to different traits colocalized within the same chromosomal region, suggesting that these represent the major QTLs affecting grain size. We carefully evaluated the QTLs associated with multiple traits, based on the physical positions of the flanking markers, and subsequently grouped the major QTLs into clusters. Collectively, three QTL clusters were identified on chromosomes 6, 7 and 11 (Table 3), Cluster 1 on chromosome 6 (9.1–19.7 Mbp) harbored three QTLs, qGL6, qGW6 and qRLW6, for GL, GW and RLW, respectively. Cluster 2 on chromosome 7 (24.1–24.8 Mbp) contained four QTLs, qUGW7, qHGW7.2, qGA7 and qGL7, associated with UGW, HGW, GA and GL. Lastly, cluster 3 on chromosome 11 (23.7–25.5 Mbp) included four QTLs, qUGW11, qHGW11, qGA11 and qGW11, related to UGW, HGW, GA and GW.
Table 3.
Grain size-related QTL clusters and putative candidate genes identified in this study.
3.4. Analysis of Genome Sequencing Data of Odae and Joun and Selection of the Putative Candidate Genes
We analyzed the whole-genome resequencing data of Odae and Joun generated previously using the Illumina HiSeq sequencing platform [32]. The raw genome sequence data of Odae (14.55 Gbp) and Joun (14.94 Gbp) were mapped to the Nipponbare reference genome sequence, resulting in 13.28-Gbp mapped data in Odae (35.58× depth) and 13.57-Gbp mapped data in Joun (36.34× depth) (Supplementary Table S2). A total of 207,540 sequence variants, including 170,039 SNPs and 37,501 InDels, were identified between Odae and Joun (Supplementary Table S3). The average variant density was 0.56 per 1 kb. The effects of variants on gene function were predicted using the SnpEff program. The variant effects were categorized into four groups, high, moderate, low and modifier, with each group containing 837 (0.4%), 6551 (3.2%), 5930 (2.9%) and 194,222 (93.6%) variants, respectively (Supplementary Table S4). Frameshift mutations (645), missense mutations (6044), synonymous mutations (4767) and upstream mutations (78,049) were the most abundant in high-impact, moderate-impact, low-impact and modifier groups, respectively.
In addition, we produced long-read sequencing data of Odae and Joun using a PacBio sequencing platform. The mean read lengths were 17,443 bp and 16,897 bp for Odae and Joun, respectively, with 22.9 Gb and 17.7 Gb of overall data for those varieties (Supplementary Table S5). The sequencing depths were 61.4× for Odae and 57.5× for Joun. Through assembly of long-read sequencing data, 107 and 648 contigs were produced with a longest contig size of 19,738,146 bp and 20,941,718 bp for Odae and Joun, respectively (Supplementary Table S6). The N50 contig lengths were 7,272,150 bp for Odae, and 12,074,287 bp for Joun. Among the contigs, we selected the contigs located in the QTL cluster on chromosomes 7 and 11. A contig (9,202,113 bp) of Odae and a contig (17,384,363 bp) of Joun, which included the QTL cluster region on chromosome 7 completely, were found. Through comparison of these contigs using the MUMmer 3.23 program, we found 43 high and 290 moderate impact-effect sequence variants in the QTL cluster on chromosome 7. Moreover, a contig (5,004,973 bp) of Odae and a contig (16,642,611 bp) of Joun, which included the QTL cluster region on chromosome 11 completely, were found. Through comparison of these contigs, 14 high and 167 moderate impact-effect sequence variants in the QTL cluster on chromosome 11 were found.
Integrating the variants detected by Illumina and PacBio sequencing, a total of 355 high or moderate impact-effect variants were located in the QTL cluster on chromosome 7, among which 170 variants were detected by both Illumina and PacBio, 22 variants by only Illumina and 163 variants by only PacBio. Likewise, a total of 351 high or moderate impact-effect variants were located in the QTL cluster on chromosome 11, among which 120 variants were detected by both Illumina and PacBio, 170 variants by only Illumina and 61 variants by only PacBio. The detailed information of the variants is shown in Supplementary Table S7.
Next, we tried to identify putative candidate genes underlying the QTLs in the two QTL clusters on chromosomes 7 and 11 by surveying the literature and examining the sequence variations between Odae and Joun based on their whole-genome resequencing and long-read sequencing data. The interval of the QTL cluster on chromosome 6 was quite large (10.6 Mbp) and was excluded from our candidate gene search. Among the genes exhibiting sequence variations between Odae and Joun, along with a moderate to high impact (as predicted by SnpEff), we selected several genes localized within the QTL clusters on chromosomes 7 and 11 and harbored domains previously reported to be associated with grain size-related traits (Table 3 and Supplementary Table S8, Figure 3).
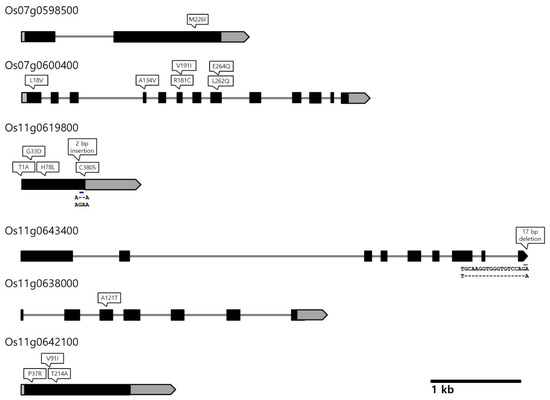
Figure 3.
Sequence variation with high or moderate impact effect in putative candidate genes identified in this study. Positions of non-synonymous SNPs and InDels are indicated above each locus. Gray boxes indicate 5’ and 3’ untranslated regions (UTRs); black boxes indicate exons; gray lines represent introns; arrowheads at the end of each locus indicate the direction of transcription.
Among the identified genes controlling the grain size in rice, only GL7/GW7/SLG7, encoding a TONNEAU1-recruiting motif protein [25], was located in the region of the clusters. It is located at 24.7 Mbp on chromosome 7, which is within the cluster 2 region. However, GL7/GW7/SLG7 showed no sequence variation between Odae and Joun; therefore, this gene was not considered a candidate for QTLs in cluster 2.
Two putative candidate genes were found in cluster 2 on chromosome 7. While Os07g0598500 (encoding a PPR domain-containing protein) contained a variation that caused a single amino acid substitution (Met to Ile) at position 226 in the encoded protein, Os07g0600400 (encoding a WD40/YVTN repeat-like domain-containing protein) contained six missense variations arising from SNPs in the coding sequence.
More putative candidate genes were found in cluster 3 on chromosome 11, including Os11g0619800 (encoding a Kelch-related domain-containing protein), Os11g0643400 (encoding an SCP family protein), Os11g0638000 (encoding GTP-binding protein engA) and Os11g0642100 (encoding a cyclin-like F-box domain-containing protein). Os11g0619800 contained four missense variations causing amino acid substitutions and a frame-shift variation caused by two base pair insertions at the 370th amino acid. The Os11g0643400 gene contained a 17 bp deletion, resulting in a frameshift variation at the 495th amino acid in exon 9. The Os11g0638000 and Os11g0642100 genes contained one and three missense variations, respectively.
4. Discussion
Rice, especially Korean japonica rice, has long been cultivated in Korea as the most important cereal grain. Growing concerns about food security due to climate change and population growth highlight the need for elite high-yielding varieties. Grain size is one of the most important target traits for improving rice yield. Therefore, a large number of candidate QTLs/genes associated with grain size regulation have been identified in rice.
Despite numerous remarkable accomplishments over the past decades, many grain size-related QTLs remain unidentified. In addition, grain size is a complex quantitative trait controlled by various genes as well as environmental factors. Therefore, identification of additional QTLs/genes is critical for a better understanding of the genetic basis of grain size-related traits. In the present study, we aimed to detect reliable QTLs affecting grain size in Korean japonica rice. To achieve this aim, we investigated six major traits related to grain size—UGW, HGW, GA, GL, GW and RLW—over two years using F8 and F9 generations of 162 RILs derived from a cross between two Korean japonica varieties with contrasting grain sizes, Odae (large grains) and Joun (small grains).
A total of 15 QTLs were detected in both F8 and F9 populations (grown in 2019 and 2020, respectively) and thus were considered as stable QTLs. More importantly, two or more QTLs linked to different traits colocalized to the same chromosomal interval; these QTLs were subsequently classified into three clusters based on the physical positions of flanking markers. Cluster 1 on chromosome 6 contained three QTLs associated with grain length, width and the ratio of length to width; cluster 2 located on chromosome 7 contained QTLs affecting grain weight, area and length; and cluster 3 found on chromosome 11 contained QTLs related to grain weight, area and width (Table 3). We, therefore, paid special attention to these three QTL clusters to mine putative candidate genes controlling grain size in Korean japonica rice. We selected a total of six putative candidate genes within the QTL clusters on chromosomes 7 and 11; these genes showed sequence variations between Odae and Joun, with moderate to high impact effects on gene function. Notably, the corresponding gene families of the candidate genes have been reported to directly or indirectly regulate grain length, grain size, development and filling by influencing various cellular process such as cell division, cell proliferation, cell expansion and mitochondrial gene expression [7,26,27].
Cluster 2 was identified at 24.1–24.7 Mbp on chromosome 7, and two putative candidate genes were found in this region. One of these, Os07g0598500 (encoding a PPR domain-containing protein), may control plant growth and development by regulating gene transcription. In maize (Zea mays), significant progress has been made toward understanding the factors contributing to grain size. For instance, mutants of a nuclear-encoded mitochondrial PPR protein gene, emp4, exhibited an extremely small endosperm size, which is highly correlated with seed size [51]. More recently, the qKW9 QTL, associated with maize kernel weight, was mapped and cloned as a PLS-DYW PPR protein-coding gene involved in C-to-U editing of ndhB, a subunit of the chloroplast NADH dehydrogenase-like complex. In the qkw9 null mutant, photosynthesis was reduced, which decreased the maternal photosynthates available for grain filling, leading to significant reduction in ear and kernel size [52]. Another gene, Os07g0600400, which encodes a WD40/YVTN repeat-like domain-containing protein, may modulate organ size in plants. Analysis of the gain and loss of function of transgenic rice plants revealed that OsWD1, a member of the WD-40 family, positively regulates seed size by enhancing the expression of GA-inducible genes, including OsEP3A and α-amylase [53]. Moreover, in cucumber (Cucumis sativus), the LITTLELEAF locus, which encodes a WD40 protein, exhibits pleiotropic effects on seed size and lateral branch number [54]. Based on these findings, we speculate that the two putative candidate genes identified on chromosome 7 of rice might be associated with regulating grain weight, area and length.
Several promising putative candidate genes, including Os11g0619800, Os11g0643400, Os11g0638000 and Os11g0642100, were identified within cluster 3 on chromosome 11. The Os11g0619800 gene encodes a Kelch-related domain-containing protein and belongs to the same gene family as the previously reported grain weight and length-related rice QTL GL3/qGL3.1 [14]. According to Zhang et al., qGL3.1 encodes a Kelch-like repeat domain-containing serine/threonine phosphatase OsPPLK1, which functions as a negative regulator of grain length, filling and weight by affecting cell proliferation [15]. The Os11g0643400 gene encodes an SCP family protein that mediates BR signaling. In rice, the SCP protein GS5 positively regulates grain size by promoting cell division and enlargement, leading to enhanced latitudinal growth in the grain [12]. In Arabidopsis, overexpression of the SCP gene increases the carpel number and seed size [55]. The Os11g0638000 gene encodes a GTP-binding protein, which has been strongly suggested to regulate grain size [8,9]. For instance, rice G-protein γ subunits GS3 and DEP1 negatively influence seed length and weight by restricting cell proliferation [56]. The fourth candidate gene, Os11g0642100, encodes a cyclin-like F-box domain-containing protein that belongs to the E3 ubiquitin ligase family of proteins, which are involved in diverse biological processes including seed development. In rice, a loss-of-function mutation of the GW2 gene, which encodes a RING-type E3 ubiquitin ligase, enhances grain width, weight and yield by affecting the rate of cell division in the spikelet hull [10].
Notably, the functions of some genes mentioned above have not yet been characterized in rice, but it is widely accepted that orthologs of these genes in different plant species, as well as in other organisms, perform similar biological functions. Our findings showed that genes regulating seed size-related traits were highly concentrated within the different QTL clusters, suggesting the importance of these clusters as primary targets for marker-assisted breeding. We strongly believe that the potential candidate genes identified in the present study will provide valuable genetic information for selecting the target genes underlying desirable traits, and will facilitate molecular breeding for improving the grain yield of rice, especially Korean japonica rice. Further studies are needed to validate the function of the putative candidate genes and to investigate the genetic correlations between traits by distinguishing the pleiotropic effects of a single gene from those of tightly linked loci affecting QTL colocalization within a specific cluster.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/agriculture12010051/s1, Figure S1: Flowchart depicting the high-throughput image analysis procedure used in this study for measuring the grain size in rice, Table S1: The physical location of the 248 KASP markers used in this study, Table S2: Summary of whole-genome resequencing data of rice varieties Odae and Joun, Table S3: Number of variants per chromosome, Table S4: Classification of variants by their effects, Table S5: Summary of whole-genome long-read sequencing data produced by PacBio platform, Table S6: Assembly summary of whole-genome long-read sequencing data produced by PacBio platform, Table S7: List of sequence variations detected in the QTL clusters on chromosomes 7 and 11, Table S8: Information about variants with high or moderate impact effect in the putative candidate genes.
Author Contributions
Conceptualization, H.J. and K.-H.K.; software, J.B.; validation, I.S.Y. and Y.-S.C.; formal analysis, C.L. and H.O.; investigation, Y.S. and K.-S.C.; resources, Y.J.W., G.-S.L. and S.L.K.; data curation, Y.S. and H.J.; writing—original draft preparation, Y.S. and H.J.; writing—review and editing, H.J.; project administration, K.-H.K. and H.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the BioGreen21 Agri-Tech Innovation Program (project code: PJ01579402), Rural Development Administration, Korea.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muthayya, S.; Sugimoto, J.D.; Montgomery, S.; Maberly, G.F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 2014, 1324, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.B.; Huang, Y.; Huang, M.; Yao, Y.; Bassu, S.; Ciais, P.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326–9331. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.K. Impact of climate change on agriculture production and its sustainable solutions. Environ. Sustain. 2019, 2, 95–96. [Google Scholar] [CrossRef]
- Sakamoto, T.; Matsuoka, M. Identifying and exploiting grain yield genes in rice. Curr. Opin. Plant Biol. 2008, 11, 209–214. [Google Scholar] [CrossRef]
- Lee, C.M.; Park, J.; Kim, B.; Seo, J.; Lee, G.; Jang, S.; Koh, H.J. Influence of Multi-Gene Allele Combinations on Grain Size of Rice and Development of a Regression Equation Model to Predict Grain Parameters. Rice 2015, 8, 33. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, R.; Jiang, L.; Zheng, J.; Wang, T.; Wang, H.; Huang, Y.; Hong, Z. Genetic bases of rice grain shape: So many genes, so little known. Trends Plant Sci. 2013, 18, 218–226. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Duan, P.; Li, Y. Control of grain size in rice. Plant Reprod. 2018, 31, 237–251. [Google Scholar] [CrossRef]
- Fan, C.; Xing, Y.; Mao, H.; Lu, T.; Han, B.; Xu, C.; Li, X.; Zhang, Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 2006, 112, 1164–1171. [Google Scholar] [CrossRef]
- Mao, H.; Sun, S.; Yao, J.; Wang, C.; Yu, S.; Xu, C.; Li, X.; Zhang, Q. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. USA 2010, 107, 19579–19584. [Google Scholar] [CrossRef]
- Song, X.J.; Huang, W.; Shi, M.; Zhu, M.Z.; Lin, H.X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef]
- Huang, K.; Wang, D.; Duan, P.; Zhang, B.; Xu, R.; Li, N.; Li, Y. WIDE AND THICK GRAIN 1, which encodes an otubain-like protease with deubiquitination activity, influences grain size and shape in rice. Plant J. 2017, 91, 849–860. [Google Scholar] [CrossRef]
- Li, Y.; Fan, C.; Xing, Y.; Jiang, Y.; Luo, L.; Sun, L.; Shao, D.; Xu, C.; Li, X.; Xiao, J.; et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011, 43, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Ni, S.; Wang, J.; Zhang, B.; Xu, R.; Wang, Y.; Chen, H.; Zhu, X.; Li, Y. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2015, 2, 15203. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Lin, Y.S.; Song, X.J.; Shen, J.B.; Huang, W.; Shan, J.X.; Zhu, M.Z.; Jiang, L.; Gao, J.P.; Lin, H.X. The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1;3. Cell Res. 2012, 22, 1666–1680. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Huang, J.; Lan, H.; Wang, C.; Yin, C.; Wu, Y.; Tang, H.; Qian, Q.; Li, J.; et al. Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc. Natl. Acad. Sci. USA 2012, 109, 21534–21539. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Gu, S.; Wan, X.; Gao, H.; Guo, T.; Su, N.; Lei, C.; Zhang, X.; Cheng, Z.; Guo, X.; et al. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 2008, 18, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, J.; Zheng, X.; Wu, F.; Lin, Q.; Heng, Y.; Tian, P.; Cheng, Z.; Yu, X.; Zhou, K.; et al. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 2017, 3, 17043. [Google Scholar] [CrossRef]
- Hu, Z.; Lu, S.J.; Wang, M.J.; He, H.; Sun, L.; Wang, H.; Liu, X.H.; Jiang, L.; Sun, J.L.; Xin, X.; et al. A Novel QTL qTGW3 Encodes the GSK3/SHAGGY-Like Kinase OsGSK5/OsSK41 that Interacts with OsARF4 to Negatively Regulate Grain Size and Weight in Rice. Mol. Plant 2018, 11, 736–749. [Google Scholar] [CrossRef]
- Ishimaru, K.; Hirotsu, N.; Madoka, Y.; Murakami, N.; Hara, N.; Onodera, H.; Kashiwagi, T.; Ujiie, K.; Shimizu, B.; Onishi, A.; et al. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 2013, 45, 707–711. [Google Scholar] [CrossRef]
- Shi, C.L.; Dong, N.Q.; Guo, T.; Ye, W.W.; Shan, J.X.; Lin, H.X. A quantitative trait locus GW6 controls rice grain size and yield through the gibberellin pathway. Plant J. 2020, 103, 1174–1188. [Google Scholar] [CrossRef]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef]
- Qian, Q. Gene Network of Grain Size and Number in Rice. In Rice Genomics, Genetics and Breeding; Springer: Berlin/Heidelberg, Germany, 2018; pp. 191–206. [Google Scholar]
- Wang, Y.; Xiong, G.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.; Zeng, L.; Xu, E.; Xu, J.; et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, R.; Li, Y. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Li, J. Molecular genetic dissection of quantitative trait loci regulating rice grain size. Annu. Rev. Genet. 2014, 48, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef]
- Hori, K.; Yamamoto, T.; Yano, M. Genetic dissection of agronomically important traits in closely related temperate japonica rice cultivars. Breed. Sci. 2017, 67, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, D.; Bohra, A.; Thudi, M.; Varshney, R.K. Fine mapping and gene cloning in the post-NGS era: Advances and prospects. Theor. Appl. Genet. 2020, 133, 1791–1810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.L.; Grondin, A.; Courtois, B.; Gantet, P. Next-Generation Sequencing Accelerates Crop Gene Discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Baek, J.; Cho, Y.-I.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Won, Y.J.; Kang, D.-Y.; Oh, H.; Kim, S.L.; et al. Single Nucleotide Polymorphism (SNP) Discovery and Kompetitive Allele-Specific PCR (KASP) Marker Development with Korean japonica Rice Varieties. Plant Breed. Biotechnol. 2018, 6, 391–403. [Google Scholar] [CrossRef]
- Cheon, K.S.; Jeong, Y.M.; Oh, H.; Oh, J.; Kang, D.Y.; Kim, N.; Lee, E.; Baek, J.; Kim, S.L.; Choi, I.; et al. Development of 454 New Kompetitive Allele-Specific PCR (KASP) Markers for Temperate japonica Rice Varieties. Plants 2020, 9, 1531. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Kang, D.-Y.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; Baek, J.; et al. Kompetitive Allele-Specific PCR Marker Development and Quantitative Trait Locus Mapping for Bakanae Disease Resistance in Korean japonica Rice Varieties. Plant Breed. Biotechnol. 2019, 7, 208–219. [Google Scholar] [CrossRef]
- Kang, D.Y.; Cheon, K.S.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; Choi, I.; Baek, J.; Kim, K.H.; et al. Rice Genome Resequencing Reveals a Major Quantitative Trait Locus for Resistance to Bakanae Disease Caused by Fusarium fujikuroi. Int. J. Mol. Sci. 2019, 20, 2598. [Google Scholar] [CrossRef]
- Cheon, K.S.; Won, Y.J.; Jeong, Y.M.; Lee, Y.Y.; Kang, D.Y.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; et al. QTL mapping for pre-harvest sprouting resistance in japonica rice varieties utilizing genome re-sequencing. Mol. Genet. Genom. 2020, 295, 1129–1140. [Google Scholar] [CrossRef]
- Baek, J.; Lee, E.; Kim, N.; Kim, S.L.; Choi, I.; Ji, H.; Chung, Y.S.; Choi, M.S.; Moon, J.K.; Kim, K.H. High Throughput Phenotyping for Various Traits on Soybean Seeds Using Image Analysis. Sensors 2020, 20, 248. [Google Scholar] [CrossRef] [PubMed]
- Lorieux, M. MapDisto: Fast and efficient computation of genetic linkage maps. Mol. Breed. 2012, 30, 1231–1235. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Basten, C.J.; Weir, B.S.; Zeng, Z.-B. QTL Cartographer: A Reference Manual and Tutorial for QTL Mapping; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 1996. [Google Scholar]
- Kumagai, M.; Nishikawa, D.; Kawahara, Y.; Wakimoto, H.; Itoh, R.; Tabei, N.; Tanaka, T.; Itoh, T. TASUKE+: A web-based platform for exploring GWAS results and large-scale resequencing data. DNA Res. 2020, 27, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol. 2013, 54, e6. [Google Scholar] [CrossRef]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Marcos, J.F.; Dal Pra, M.; Giulini, A.; Costa, L.M.; Gavazzi, G.; Cordelier, S.; Sellam, O.; Tatout, C.; Paul, W.; Perez, P.; et al. empty pericarp4 encodes a mitochondrion-targeted pentatricopeptide repeat protein necessary for seed development and plant growth in maize. Plant Cell 2007, 19, 196–210. [Google Scholar] [CrossRef]
- Huang, J.; Lu, G.; Liu, L.; Raihan, M.S.; Xu, J.; Jian, L.; Zhao, L.; Tran, T.M.; Zhang, Q.; Liu, J.; et al. The Kernel Size-Related Quantitative Trait Locus qKW9 Encodes a Pentatricopeptide Repeat Protein That Aaffects Photosynthesis and Grain Filling. Plant Physiol. 2020, 183, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- He, S.-L. Functional Characterization of a WD-Repeat Protein Gene (OsWD1) in Rice. Am. J. Agric. For. 2018, 6, 18–27. [Google Scholar] [CrossRef]
- Yang, L.; Liu, H.; Zhao, J.; Pan, Y.; Cheng, S.; Lietzow, C.D.; Wen, C.; Zhang, X.; Weng, Y. LITTLELEAF (LL) encodes a WD40 repeat domain-containing protein associated with organ size variation in cucumber. Plant J. 2018, 95, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Li, J.; Walker, J.C. Overexpression of a serine carboxypeptidase increases carpel number and seed production in Arabidopsis thaliana. Food Energy Secur. 2012, 1, 61–69. [Google Scholar] [CrossRef]
- Li, S.; Liu, W.; Zhang, X.; Liu, Y.; Li, N.; Li, Y. Roles of the Arabidopsis G protein gamma subunit AGG3 and its rice homologs GS3 and DEP1 in seed and organ size control. Plant Signal. Behav. 2012, 7, 1357–1359. [Google Scholar] [CrossRef] [PubMed][Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).