
Sequencing of the Whole Genome of a Bacterium of the Genus Achromobacter Reveals Its Potential for Xenobiotics Biodegradation



Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Culture, Genomic DNA Isolation, and Sequencing
2.2. Genome Assembly and Annotation, Phylogenetic Analysis, Comparative Genome Analysis
3. Results and Discussion
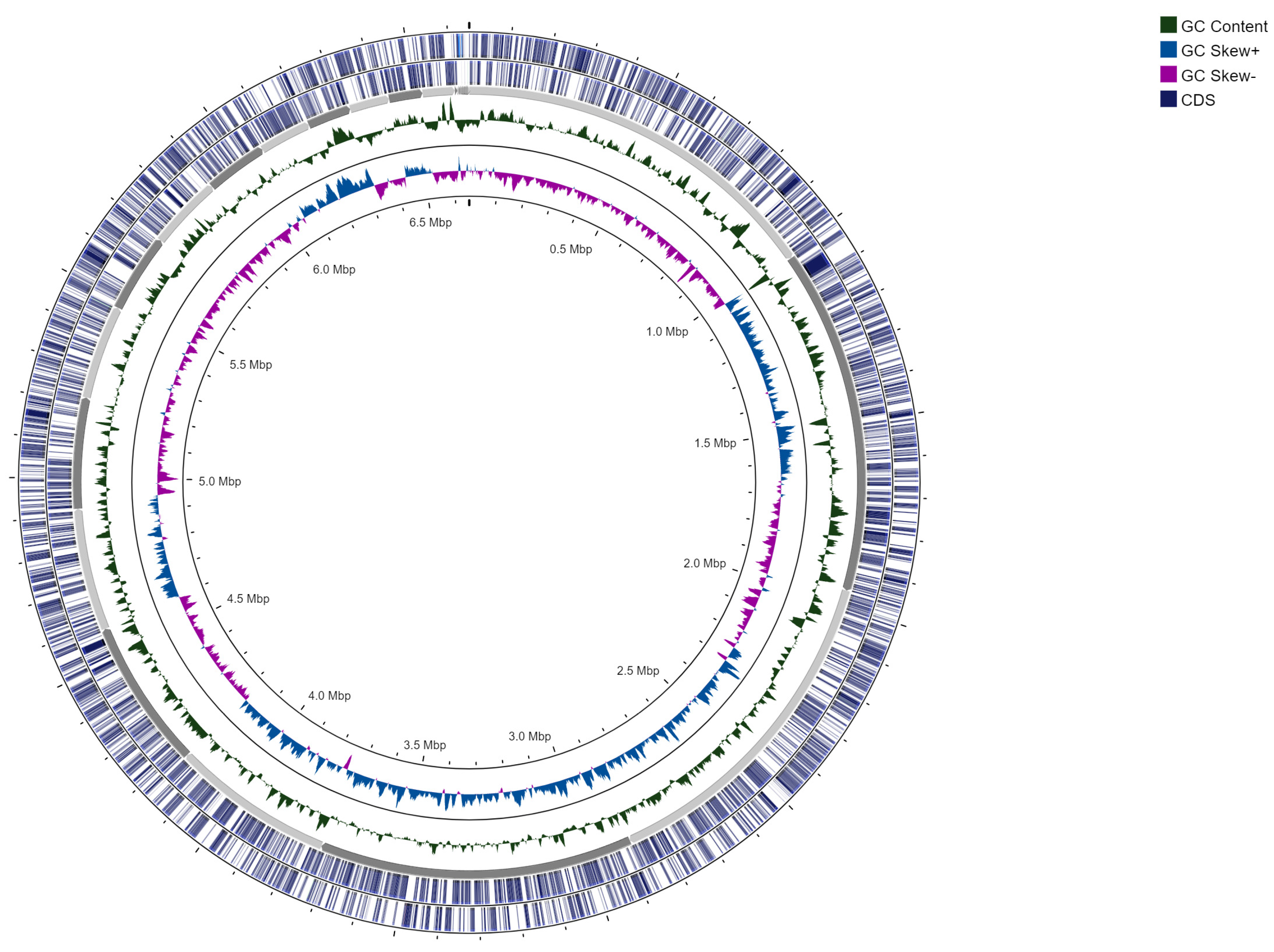
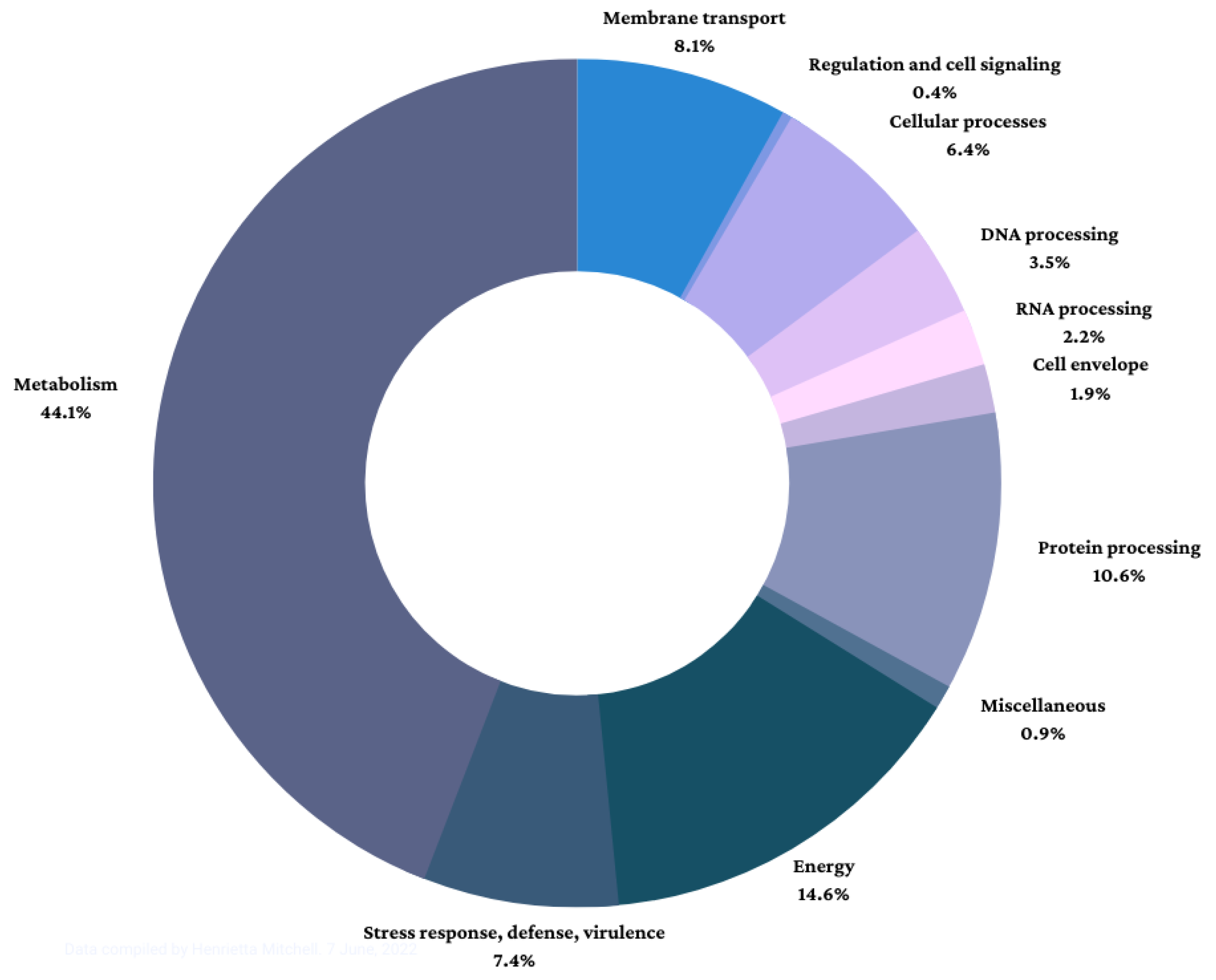
3.1. Genome Assembly and Annotation
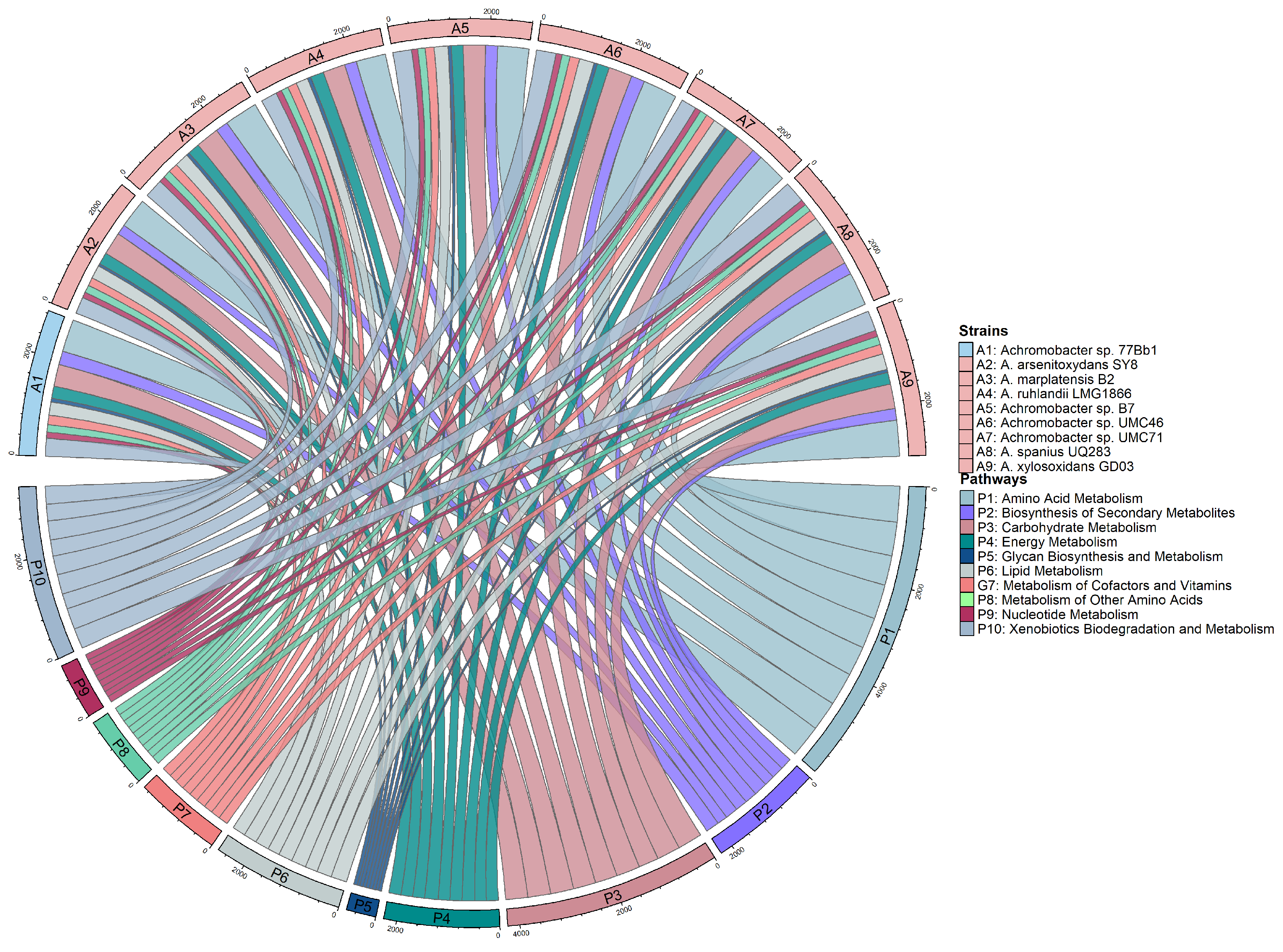
3.2. Analysis of Genes from Different Metabolic Pathways
3.3. Analysis of Genes Taking Part in Biosynthesis of Secondary Metabolites
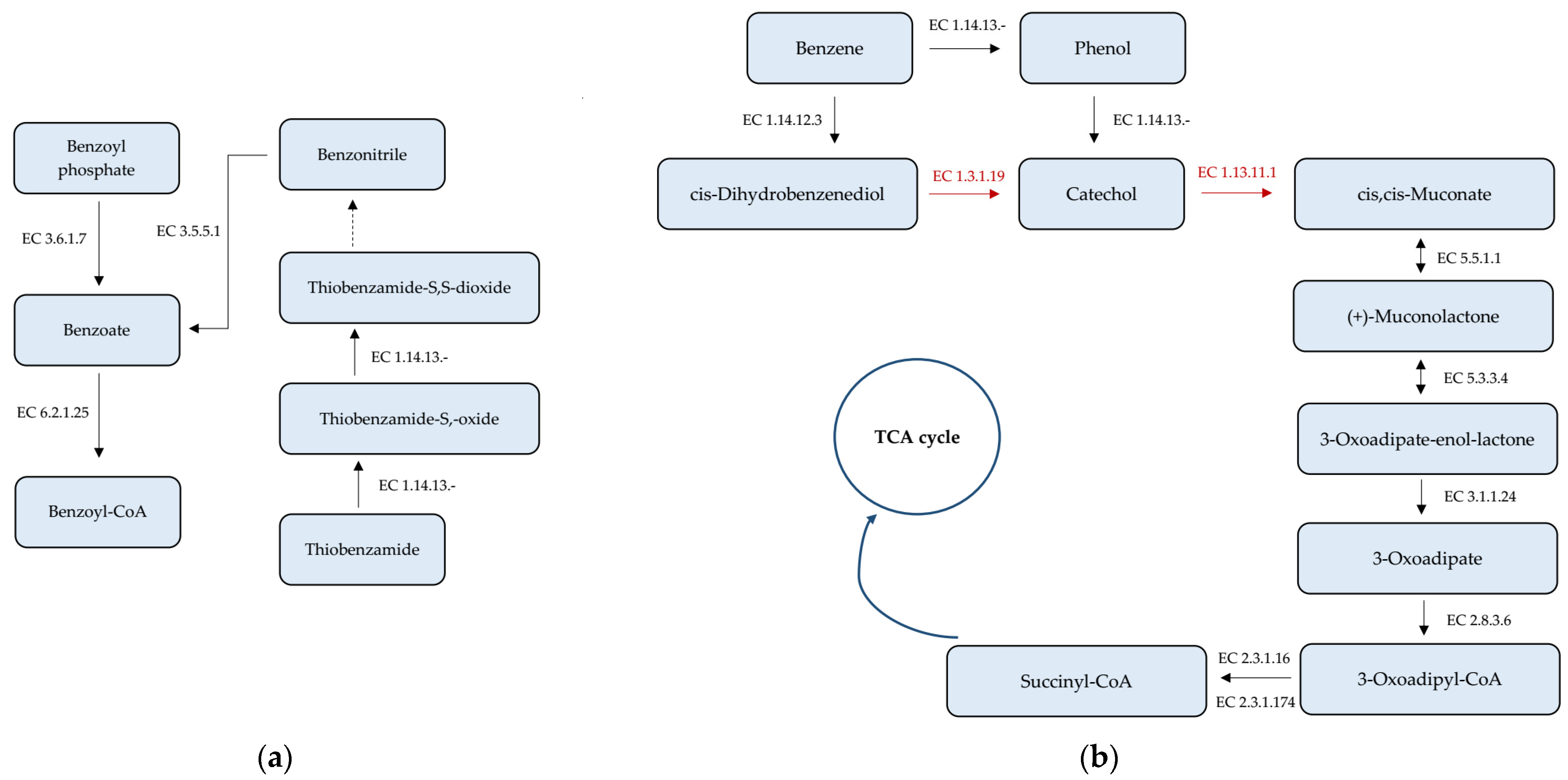
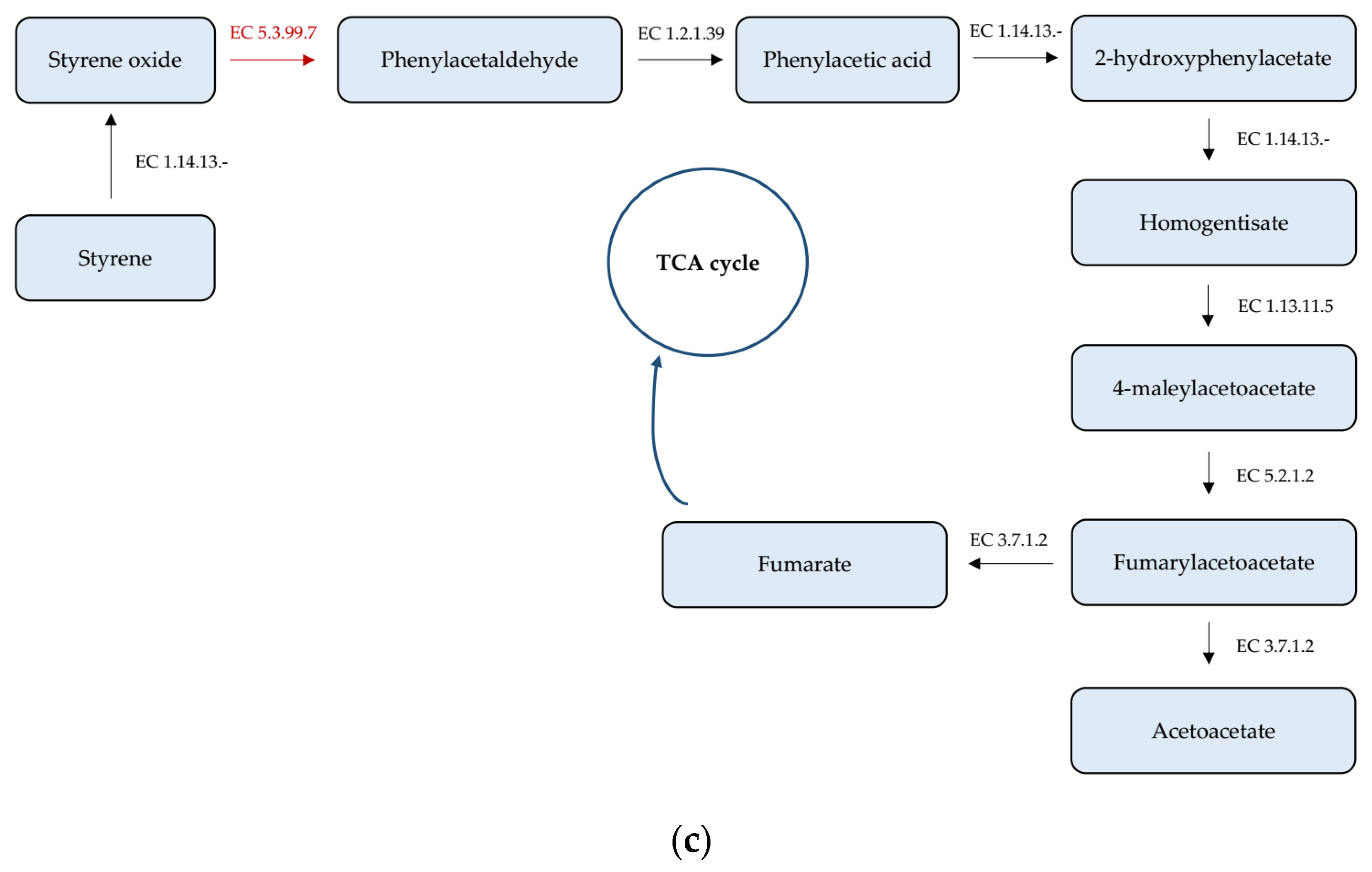
3.4. Analysis of Genes Taking Part in Xenobiotics Biodegradation and Metabolism
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kuklinsky-Sobral, J.; Araújo, W.L.; Mendes, R.; Geraldi, I.O.; Pizzirani-Kleiner, A.A.; Azevedo, J.L. Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environ. Microbiol. 2004, 6, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Aleklett, K.; Leff, J.W.; Fierer, N.; Hart, M. Wild plant species growing closely connected in a subalpine meadow host distinct root-associated bacterial communities. PeerJ 2015, 2015, e804. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, A.S.; Thijs, S.; Beckers, B.; González-Chávez, M.C.; Weyens, N.; Carrillo-González, R.; Vangronsveld, J. Community structure and diversity of endophytic bacteria in seeds of three consecutive generations of Crotalaria pumila growing on metal mine residues. Plant Soil 2018, 422, 51–66. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Ver Loren van Themaat, E.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Gaiero, J.R.; McCall, C.A.; Thompson, K.A.; Day, N.J.; Best, A.S.; Dunfield, K.E. Inside the root microbiome: Bacterial root endophytes and plant growth promotion. Am. J. Bot. 2013, 100, 1738–1750. [Google Scholar] [CrossRef]
- Santoyo, G.; Moreno-Hagelsieb, G.; del Carmen Orozco-Mosqueda, M.; Glick, B.R. Plant growth-promoting bacterial endophytes. Microbiol. Res. 2016, 183, 92–99. [Google Scholar] [CrossRef]
- Nannipieri, P.; Ascher, J.; Ceccherini, M.T.; Landi, L.; Pietramellara, G.; Renella, G. Microbial diversity and soil functions. Eur. J. Soil Sci. 2017, 68, 12–26. [Google Scholar] [CrossRef]
- De Maayer, P.; Chan, W.Y.; Venter, S.N.; Toth, I.K.; Birch, P.R.J.; Joubert, F.; Coutinho, T.A. Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J. Bacteriol. 2010, 192, 2936–2937. [Google Scholar] [CrossRef]
- Kahlke, T.; Goesmann, A.; Hjerde, E.; Willassen, N.P.; Haugen, P. Unique core genomes of the bacterial family vibrionaceae: Insights into niche adaptation and speciation. BMC Genom. 2012, 13, 179–190. [Google Scholar] [CrossRef]
- Lo, K.J.; Lin, S.S.; Lu, C.W.; Kuo, C.H.; Liu, C. Te Whole-genome sequencing and comparative analysis of two plant-associated strains of Rhodopseudomonas palustris (PS3 and YSC3). Sci. Rep. 2018, 8, 12769. [Google Scholar] [CrossRef]
- Azarbaijani, R.; Yeganeh, L.P.; Blom, J.; Younesi, H.; Fazeli, S.A.S.; Tabatabaei, M.; Salekdeh, G.H. Comparative genome analysis of Oceanimonas sp. GK1, a halotolerant bacterium with considerable xenobiotics degradation potentials. Ann. Microbiol. 2016, 66, 703–716. [Google Scholar] [CrossRef]
- Torres-Cortés, G.; Millán, V.; Ramírez-Saad, H.C.; Nisa-Martínez, R.; Toro, N.; Martínez-Abarca, F. Characterization of novel antibiotic resistance genes identified by functional metagenomics on soil samples. Environ. Microbiol. 2011, 13, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef]
- Şahin, A.D.; Saçan, M.T. Understanding the toxic potencies of xenobiotics inducing TCDD/TCDF-like effects. SAR QSAR Environ. Res. 2018, 29, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R. Biodegradation of xenobiotics- a way for environmental detoxification. Int. J. Dev. Res. 2017, 7, 14082–14087. [Google Scholar]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Jiménez-Vázquez, K.R.; García-Cárdenas, E.; Barrera-Ortiz, S.; Ortiz-Castro, R.; Ruiz-Herrera, L.F.; Ramos-Acosta, B.P.; Coria-Arellano, J.L.; Sáenz-Mata, J.; López-Bucio, J. The plant beneficial rhizobacterium Achromobacter sp. 5B1 influences root development through auxin signaling and redistribution. Plant J. 2020, 103, 1639–1654. [Google Scholar] [CrossRef]
- Jana, G.A.; Yaish, M.W. Genome analysis of a salinity adapted Achromobacter xylosoxidans rhizobacteria from the date palm. Rhizosphere 2021, 19, 100401–100412. [Google Scholar] [CrossRef]
- Wass, T.J.; Syed-Ab-Rahman, S.F.; Carvalhais, L.C.; Ferguson, B.J.; Schenk, P.M. Complete Genome Sequence of Achromobacter spanius UQ283, a Soilborne Isolate Exhibiting Plant Growth-Promoting Properties. Microbiol. Resour. Announc. 2019, 8, e00236-19. [Google Scholar] [CrossRef]
- Iyer, R.; Damania, A. Draft genome sequence of the broad-spectrum xenobiotic degrader Achromobacter xylosoxidans ADAF13. Genome Announc. 2016, 4, e00203-16. [Google Scholar] [CrossRef]
- Mishra, S.; Lin, Z.; Pang, S.; Zhang, W.; Bhatt, P.; Chen, S. Recent Advanced Technologies for the Characterization of Xenobiotic-Degrading Microorganisms and Microbial Communities. Front. Bioeng. Biotechnol. 2021, 9, 632059. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Ye, C.C.; Zhou, Q.Z.; Wu, X.Y.; Yuan, J.P.; Peng, J.; Deng, H.; Wang, J.H. Genome sequencing reveals the potential of Achromobacter sp. HZ01 for bioremediation. Front. Microbiol. 2017, 8, 1507. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Rensing, C.; Li, X.; Wang, G. Novel gene clusters involved in arsenite oxidation and resistance in two arsenite oxidizers: Achromobacter sp. SY8 and Pseudomonas sp. TS44. Appl. Microbiol. Biotechnol. 2009, 4, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Hayase, N.; Taira, K.; Tomizuka, N. Molecular relationship of chromosomal genes encoding biphenyl/poychlorinated biphenyl catabolism: Some soil bacteria possess a highly conserved bph operon. J. Bacteriol. 1989, 171, 5467–5472. [Google Scholar] [CrossRef] [PubMed]
- Jencova, V.; Strnad, H.; Chodora, Z.; Ulbrich, P.; Vlcek, C.; Hickey, W.J.; Paces, V. Nucleotide sequence, organization and characterization of the (halo)aromatic acid catabolic plasmid pA81 from Achromobacter xylosoxidans A8. Res. Microbiol. 2008, 159, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.C.; Li, J.; Liang, F.R.; Yi, M.; Xu, X.M.; Yuan, J.P.; Peng, J.; Wu, C.F.; Wang, J.H. Isolation and characterization of a novel hydrocarbon-degrading bacterium Achromobacter sp. HZ01 from the crude oil-contaminated seawater at the Daya Bay, southern China. Mar. Pollut. Bull. 2014, 83, 79–86. [Google Scholar] [CrossRef]
- Andrews, S. FastQC—A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 February 2023).
- Krueger, F. Trim Galore!: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 3 February 2023).
- Marcel, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2013, 17, 10–12. [Google Scholar]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, 431–433. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Gerdes, S.; Olsen, G.J.; Olson, R.; Pusch, G.D.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Yoo, H. PATtyFams: Protein families for the microbial genomes in the PATRIC database. Front. Microbiol. 2016, 7, 118–129. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132–145. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Stevens, R.; Scheuermann, R.H. The Bacterial and Viral Bioinformatics Resource Center (BV-BRC). Available online: https://www.bv-brc.org/ (accessed on 3 February 2023).
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Mining secondary metabolism of Achromobacter and analysis of key genes of petroleum degradation. IOP Conf. Ser. Earth Environ. Sci. 2021, 692, 042032. [Google Scholar] [CrossRef]
- Schwengers, O.; Hoek, A.; Fritzenwanker, M.; Falgenhauer, L.; Hain, T.; Chakraborty, T.; Goesmann, A. ASA3P: An automatic and scalable pipeline for the assembly, annotation and higher-level analysis of closely related bacterial isolates. PLoS Comput. Biol. 2020, 16, e1007134. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 3, 7–14. [Google Scholar] [CrossRef]
- Integrated Microbial Genomes and Microbiomes Achromobacter sp. UMC46. Available online: https://img.jgi.doe.gov/cgi-bin/m/main.cgi?section=TaxonDetail&page=taxonDetail&taxon_oid=2921333427 (accessed on 3 February 2023).
- Li, X.; Hu, Y.; Gong, J.; Lin, Y.; Johnstone, L.; Rensing, C.; Wang, G. Genome sequence of the highly efficient arsenite-oxidizing bacterium Achromobacter arsenitoxydans SY8. J. Bacteriol. 2012, 194, 1243–1244. [Google Scholar] [CrossRef]
- Syed-Ab-Rahman, S.F.; Carvalhais, L.C.; Chua, E.; Xiao, Y.; Wass, T.J.; Schenk, P.M. Identification of soil bacterial isolates suppressing different phytophthora spp. And promoting plant growth. Front. Plant Sci. 2018, 9, 1502. [Google Scholar] [CrossRef]
- Sriram, R.; Sun, J.; Villanueva-Meyer, J.; Mutch, C.; De Los Santos, J.; Peters, J.; Korenchan, D.E.; Neumann, K.; Van Criekinge, M.; Kurhanewicz, J.; et al. Detection of Bacteria-Specific Metabolism Using Hyperpolarized [2-13C]Pyruvate. ACS Infect. Dis. 2018, 11, 797–805. [Google Scholar] [CrossRef]
- Liu, S.; Chen, Q.; Ma, T.; Wang, M.; Ni, J. Genomic insights into metabolic potentials of two simultaneous aerobic denitrification and phosphorus removal bacteria, Achromobacter sp. GAD3 and Agrobacterium sp. LAD9. FEMS Microbiol. Ecol. 2018, 1, fiy020. [Google Scholar] [CrossRef]
- Mohapatra, B.; Kazy, S.K.; Sar, P. Comparative genome analysis of arsenic reducing, hydrocarbon metabolizing groundwater bacterium Achromobacter sp. KAs 3-5T explains its competitive edge for survival in aquifer environment. Genomics 2019, 111, 1604–1619. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Al-Harrasi, A.; Lee, I.J. Complete genome sequencing and analysis of endophytic Sphingomonas sp. LK11 and its potential in plant growth. 3 Biotech 2018, 8, 389. [Google Scholar] [CrossRef]
- Salehizadeh, H.; Yan, N.; Farnood, R. Recent advances in microbial CO2 fixation and conversion to value-added products. Chem. Eng. J. 2020, 390, 124584–124602. [Google Scholar] [CrossRef]
- Sharrar, A.M.; Crits-Christoph, A.; Méheust, R.; Diamond, S.; Starr, E.P.; Banfield, J.F. Bacterial secondary metabolite biosynthetic potential in soil varies with phylum, depth, and vegetation type. mBio 2020, 11, e00416-20. [Google Scholar] [CrossRef]
- Pan, J.J.; Solbiati, J.O.; Ramamoorthy, G.; Hillerich, B.S.; Seidel, R.D.; Cronan, J.E.; Almo, S.C.; Poulter, C.D. Biosynthesis of squalene from farnesyl diphosphate in Bacteria: Three steps catalyzed by three enzymes. ACS Cent. Sci. 2015, 1, 77–82. [Google Scholar] [CrossRef]
- Curlango-Rivera, G.; Gunawardena, U.; Wen, F.; Zhao, X.; Xiong, Z.; Hawes, M.C. Roots: Contribution to the Rhizosphere. eLS 2016, 1, 1–7. [Google Scholar]
- Marmulla, R.; Harder, J. Microbial monoterpene transformations-a review. Front. Microbiol. 2014, 5, 346–359. [Google Scholar] [CrossRef]
- Zhang, L.L.; Fan, G.; Li, X.; Ren, J.N.; Huang, W.; Pan, S.Y.; He, J. Identification of functional genes associated with the biotransformation of limonene to trans-dihydrocarvone in Klebsiella sp. O852. J. Sci. Food Agric. 2022, 102, 3297–3307. [Google Scholar] [CrossRef] [PubMed]
- Soares-Castro, P.; Soares, F.; Santos, P.M. Current advances in the bacterial toolbox for the biotechnological production of monoterpene-based aroma compounds. Molecules 2021, 26, 91. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.S.; Vestergaard, J.M.; D’Amore, F.; Gørløv, J.S.; Toft, G.; Ramlau-Hansen, C.H.; Stokholm, Z.A.; Iversen, I.B.; Nissen, M.S.; Kolstad, H.A. Styrene exposure and risk of lymphohematopoietic malignancies in 73,036 reinforced plastics workers. Epidemiology 2018, 29, 342–351. [Google Scholar] [CrossRef]
- O’Leary, N.D.; O’Connor, K.E.; Dobson, A.D.W. Biochemistry, genetics and physiology of microbial styrene degradation. FEMS Microbiol. Rev. 2002, 26, 403–417. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Miranda, R.R.; Eddingsaas, N.C.; Chu, J.; Freezman, I.M.; Tyler, A.C.; Hudson, A.O. Polystyrene Degradation by Exiguobacterium sp. RIT 594: Preliminary Evidence for a Pathway Containing an Atypical Oxygenase. Microorganisms 2022, 10, 1619. [Google Scholar] [CrossRef]
- Ganesh Kumar, A.; Hinduja, M.; Sujitha, K.; Nivedha Rajan, N.; Dharani, G. Biodegradation of polystyrene by deep-sea Bacillus paralicheniformis G1 and genome analysis. Sci. Total Environ. 2021, 774, 145002–145010. [Google Scholar] [CrossRef]
- Ebciba, C.; Pavithra, N.; Chris Felshia, F.; Gnanamani, A. Exploring the styrene metabolism by aerobic bacterial isolates for the effective management of leachates in an aqueous system. RSC Adv. 2020, 44, 26535–26545. [Google Scholar]
- Cabrera, M.Á.; Márquez, S.L.; Pérez-Donoso, J.M. Comparative Genomic Analysis of Antarctic Pseudomonas Isolates with 2,4,6-Trinitrotoluene Transformation Capabilities Reveals Their Unique Features for Xenobiotics Degradation. Genes 2022, 13, 1354. [Google Scholar] [CrossRef]
- Ravintheran, S.K.; Sivaprakasam, S.; Loke, S.; Lee, S.Y.; Manickam, R.; Yahya, A.; Croft, L.; Millard, A.; Parimannan, S.; Rajandas, H. Complete genome sequence of Sphingomonas paucimobilis AIMST S2, a xenobiotic-degrading bacterium. Sci. Data 2019, 6, 280. [Google Scholar] [CrossRef]
- Parales, R.E.; Luu, R.A.; Hughes, J.G.; Ditty, J.L. Bacterial chemotaxis to xenobiotic chemicals and naturally-occurring analogs. Curr. Opin. Biotechnol. 2015, 33, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.A.; Ruiz-Manzano, A.; Rojo, F. Inactivation of cytochrome o ubiquinol oxidase relieves catabolic repression of the Pseudomonas putida GPo1 alkane degradation pathway. J. Bacteriol. 2002, 184, 3785–3793. [Google Scholar] [CrossRef]
- Kube, M.; Chernikova, T.N.; Al-Ramahi, Y.; Beloqui, A.; Lopez-Cortez, N.; Guazzaroni, M.E.; Heipieper, H.J.; Klages, S.; Kotsyurbenko, O.R.; Langer, I.; et al. Genome sequence and functional genomic analysis of the oil-degrading bacterium Oleispira antarctica. Nat. Commun. 2013, 4, 2156. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, W.; Cheng, J.; Ren, Y.; Zhao, G.; Gao, C.; Tang, Y.; Liu, X.; Han, W.; Peng, X.; et al. Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans NG80-2 isolated from a deep-subsurface oil reservoir. Proc. Natl. Acad. Sci. USA 2007, 27, 5602–5607. [Google Scholar] [CrossRef]
- Nie, Y.; Tang, Y.Q.; Li, Y.; Chi, C.Q.; Cai, M.; Wu, X.L. The genome sequence of polymorphum gilvum SL003B-26A1 T reveals its genetic basis for crude oil degradation and adaptation to the saline soil. PLoS ONE 2012, 7, e31261. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, A.V.; Morris, B.E.L.; Pereira, I.A.C.; McInerney, M.J.; Austin, R.N.; Groves, J.T.; Kukor, J.J.; Suflita, J.M.; Young, L.Y.; Zylstra, G.J.; et al. The genome sequence of Desulfatibacillum alkenivorans AK-01: A blueprint for anaerobic alkane oxidation. Environ. Microbiol. 2012, 14, 101–113. [Google Scholar] [CrossRef]
- BV-BRC 3.26.4. Available online: https://www.bv-brc.org/view/PathwayList/?eq(pathway_name,%22Drugmetabolism-otherenzymes%22)#view_tab (accessed on 3 February 2023).
- BV-BRC 3.26.4. Available online: https://www.bv-brc.org/view/PathwayList/?eq(pathway_name,%22Drugmetabolism-cytochromeP450%22)#view_tab (accessed on 3 February 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain_ID | Strain_Name | BioSample |
|---|---|---|
| A1 | Achromobacter sp. 77Bb1 | SAMN31831554 |
| A2 | Achromobacter arsenitoxydans SY8 | SAMN02469904 |
| A3 | Achromobacter marplatensis B2 | SAMN07270369 |
| A4 | Achromobacter ruhlandii LMG1866 | SAMNEA6647237 |
| A5 | Achromobacter sp. B7 | SAMN09690389 |
| A6 | Achromobacter sp. UMC46 | SAMN05245113 |
| A7 | Achromobacter sp. UMC71 | SAMN05245114 |
| A8 | Achromobacter spanius UQ283 | SAMN10643443 |
| A9 | Achromobacter xylosoxidans GD03 | SAMN15929548 |
| Attribute | Value |
|---|---|
| Genome size (bp) | 6,651,435 |
| Number of contigs | 57 |
| GC content [%] | 64.023 |
| CDS | 6026 |
| Protein function assigned | 4855 |
| Hypothetical proteins | 1171 |
| tRNA | 55 |
| Functional Superclass | No. of Genes |
|---|---|
| Cell envelope | 45 |
| Membrane transport | 195 |
| Cellular processes | 155 |
| DNA processing | 84 |
| RNA processing | 52 |
| Protein processing | 257 |
| Regulation and cell signaling | 9 |
| Energy | 352 |
| Metabolism | 1067 |
| Stress response, defense, virulence | 180 |
| Attribute | A1 | A2 | A3 | A4 | A5 | A6 | A7 | A8 | A9 |
|---|---|---|---|---|---|---|---|---|---|
| Genome size (Mbp) | 6.651 | 6.157 | 6.885 | 6.865 | 6.237 | 6.68 | 6.341 | 6.688 | 6.761 |
| Number of contigs | 57 | 105 | 61 | 86 | 1 | 1 | 1 | 1 | 1 |
| CDS | 6026 | 5646 | 6469 | 6458 | 5697 | 6238 | 5906 | 6331 | 6333 |
| Contig N50 (bp) | 855,260 | 106,744 | 471,181 | 181,399 | - | - | - | - | - |
| Functional Superclass | No. of Genes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | A4 | A5 | A6 | A7 | A8 | A9 | |
| Cell envelope | 45 | 41 | 39 | 37 | 47 | 39 | 36 | 42 | 37 |
| Membrane transport | 195 | 221 | 179 | 192 | 181 | 181 | 237 | 247 | 218 |
| Regulation and cell signaling | 9 | 9 | 19 | 9 | 9 | 11 | 9 | 20 | 9 |
| Cellular processes | 155 | 147 | 170 | 147 | 159 | 172 | 159 | 162 | 153 |
| DNA processing | 84 | 84 | 86 | 89 | 87 | 85 | 89 | 89 | 82 |
| RNA processing | 52 | 70 | 50 | 50 | 72 | 54 | 66 | 54 | 59 |
| Protein processing | 257 | 256 | 259 | 268 | 255 | 274 | 261 | 269 | 234 |
| Energy | 352 | 354 | 349 | 397 | 365 | 340 | 364 | 399 | 351 |
| Metabolism | 1067 | 938 | 998 | 1047 | 1040 | 959 | 964 | 1020 | 865 |
| Stress response, defense, virulence | 180 | 173 | 167 | 172 | 179 | 170 | 145 | 177 | 144 |
| Metabolic Pathway | No. of Proteins | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | A4 | A5 | A6 | A7 | A8 | A9 | |
| Amino acid metabolism | 672 | 617 | 687 | 613 | 642 | 712 | 616 | 694 | 742 |
| Biosynthesis of polyketides and nonribosomal peptides | 50 | 32 | 40 | 45 | 41 | 48 | 40 | 43 | 43 |
| Biosynthesis of secondary metabolites | 282 | 212 | 262 | 248 | 253 | 279 | 220 | 253 | 240 |
| Carbohydrate metabolism | 455 | 472 | 482 | 465 | 458 | 495 | 418 | 484 | 508 |
| Energy metabolism | 252 | 246 | 245 | 277 | 242 | 252 | 259 | 237 | 245 |
| Glycan biosynthesis and metabolism | 75 | 53 | 67 | 68 | 67 | 70 | 64 | 59 | 64 |
| Lipid metabolism | 272 | 278 | 287 | 249 | 288 | 307 | 258 | 309 | 330 |
| Metabolism of cofactors and vitamins | 196 | 169 | 194 | 193 | 187 | 202 | 188 | 192 | 198 |
| Nucleotide metabolism | 123 | 118 | 123 | 122 | 124 | 128 | 116 | 120 | 128 |
| Xenobiotics biodegradation and metabolism | 371 | 345 | 392 | 334 | 390 | 406 | 318 | 439 | 419 |
| Gene | Contig No. | Strand | Start Position | End Position | NA Length (bp) |
|---|---|---|---|---|---|
| Nitrilotriacetate monooxygenase component A (1.14.13-) | 17 5 | + − | 49,978 114,628 | 51,315 115,914 | 1338 1287 |
| Nitrilotriacetate monooxygenase component B (1.14.13-) | 3 4 5 7 3 1 11 | + + + + − − + | 322,411 215,673 14,315 140,548 68,595 777 7978 | 323,079 215,673 14,833 141,192 69,236 1409 8502 | 669 618 519 645 642 633 525 |
| Plant-induced nitrilase (3.5.5.1) | 4 4 | + − | 214,093 234,963 | 215,052 235,886 | 960 924 |
| Acylphosphate phosphohydrolase (3.6.1.7) | 3 | − | 329,557 | 329,865 | 309 |
| Benzoate-CoA ligase (6.2.1.25) | 9 | + | 136,740 | 138,419 | 1680 |
| Aromatic-ring-hydroxylating dioxygenase (1.14.12.3) | 3 | + | 902,075 | 902,608 | 534 |
| Muconate cycloisomerase (5.5.1.1) | 10 4 | + + | 44,014 209,116 | 45,150 210,243 | 1137 1128 |
| Muconolactone isomerase (5.3.3.4) | 8 | + | 91,217 | 91,492 | 276 |
| Beta-ketoadipate enol-lactone hydrolase (3.1.1.24) | 3 8 | + + | 719,268 91,855 | 720,092 92,634 | 825 780 |
| 3-oxoadipate CoA-transferase subunit A (2.8.3.6) | 18 3 9 1 8 | + + + − + | 5051 723,751 148,213 662,567 89,883 | 5701 724,416 148,893 663,400 90,560 | 651 666 681 834 678 |
| 3-oxoadipate CoA-transferase subunit B (2.8.3.6) | 8 9 18 3 1 | + + + + + | 90,557 148,904 5698 724,418 661,840 | 91,201 149,590 6378 725,143 662,574 | 645 687 681 726 735 |
| 3-ketoacyl-CoA thiolase (2.3.1.16) | 1 3 | − − | 974,655 415,601 | 975,839 416,782 | 1185 1182 |
| 3-oxoadipyl-CoA thiolase (2.3.1.174) | 1 | − | 282,888 | 284,093 | 1206 |
| Phenylacetaldehyde dehydrogenase (1.2.1.39) | 13 | + | 22,603 | 24,114 | 1512 |
| Homogentisate 1,2-dioxygenase (1.13.11.5) | 7 17 | − − | 269,021 36,021 | 270,319 37,328 | 1299 1308 |
| Maleylacetoacetate isomerase (5.2.1.2) | 17 4 | + + | 20,488 672,336 | 21,198 672,980 | 711 645 |
| Fumarylacetoacetase (3.7.1.2) | 7 17 | − − | 267,582 37,667 | 268,901 38,980 | 1320 1314 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzec-Grządziel, A.; Gałązka, A. Sequencing of the Whole Genome of a Bacterium of the Genus Achromobacter Reveals Its Potential for Xenobiotics Biodegradation. Agriculture 2023, 13, 1519. https://doi.org/10.3390/agriculture13081519
Marzec-Grządziel A, Gałązka A. Sequencing of the Whole Genome of a Bacterium of the Genus Achromobacter Reveals Its Potential for Xenobiotics Biodegradation. Agriculture. 2023; 13(8):1519. https://doi.org/10.3390/agriculture13081519
Chicago/Turabian StyleMarzec-Grządziel, Anna, and Anna Gałązka. 2023. "Sequencing of the Whole Genome of a Bacterium of the Genus Achromobacter Reveals Its Potential for Xenobiotics Biodegradation" Agriculture 13, no. 8: 1519. https://doi.org/10.3390/agriculture13081519
APA StyleMarzec-Grządziel, A., & Gałązka, A. (2023). Sequencing of the Whole Genome of a Bacterium of the Genus Achromobacter Reveals Its Potential for Xenobiotics Biodegradation. Agriculture, 13(8), 1519. https://doi.org/10.3390/agriculture13081519