Integrated Analysis Reveals Genetic Basis of Growth Curve Parameters in an F2 Designed Pig Population Based on Genome and Transcriptome Data

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal and Genotyping Data
2.3. Animals and Transcriptome Data
2.4. Growth Curve Construction
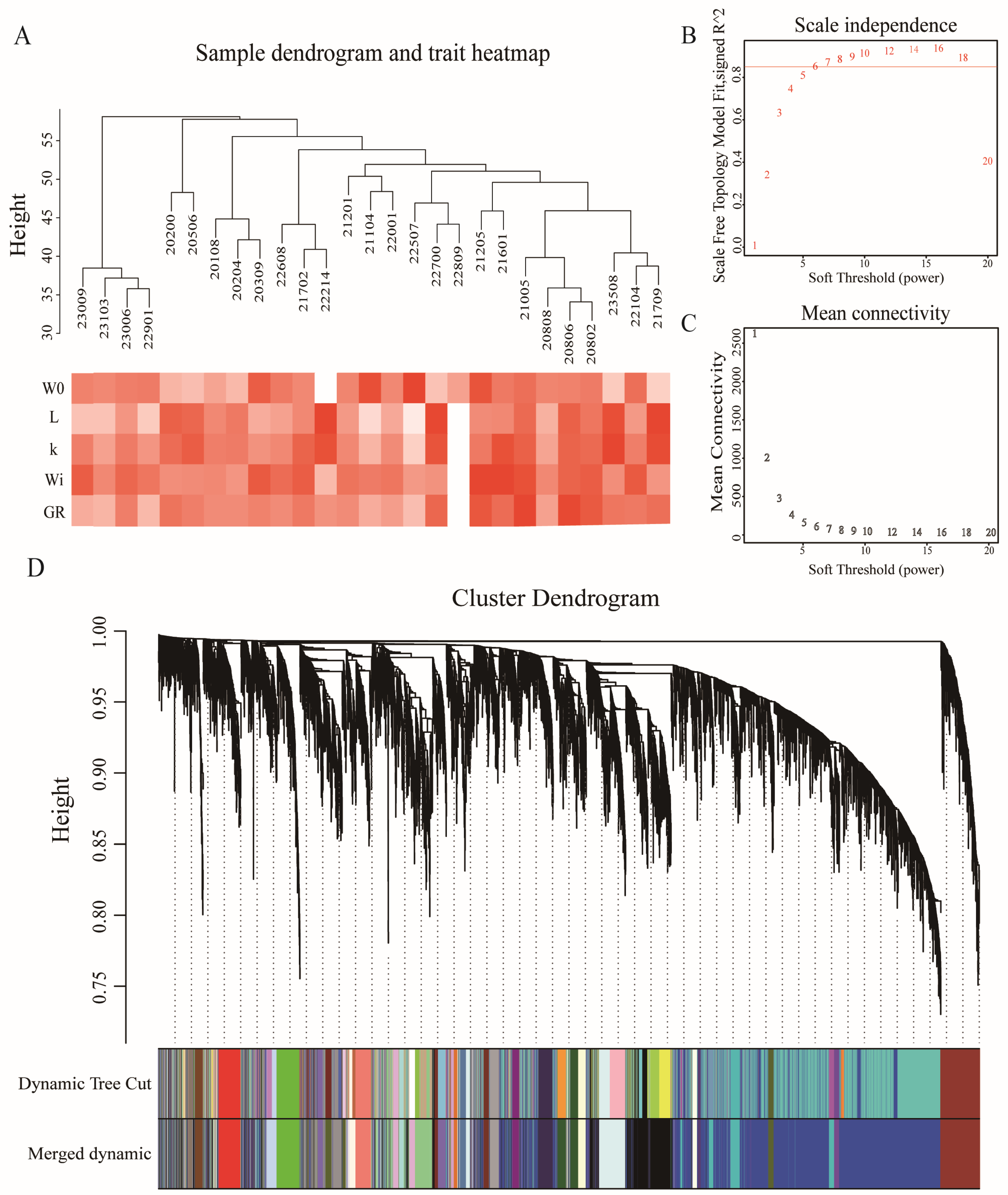
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
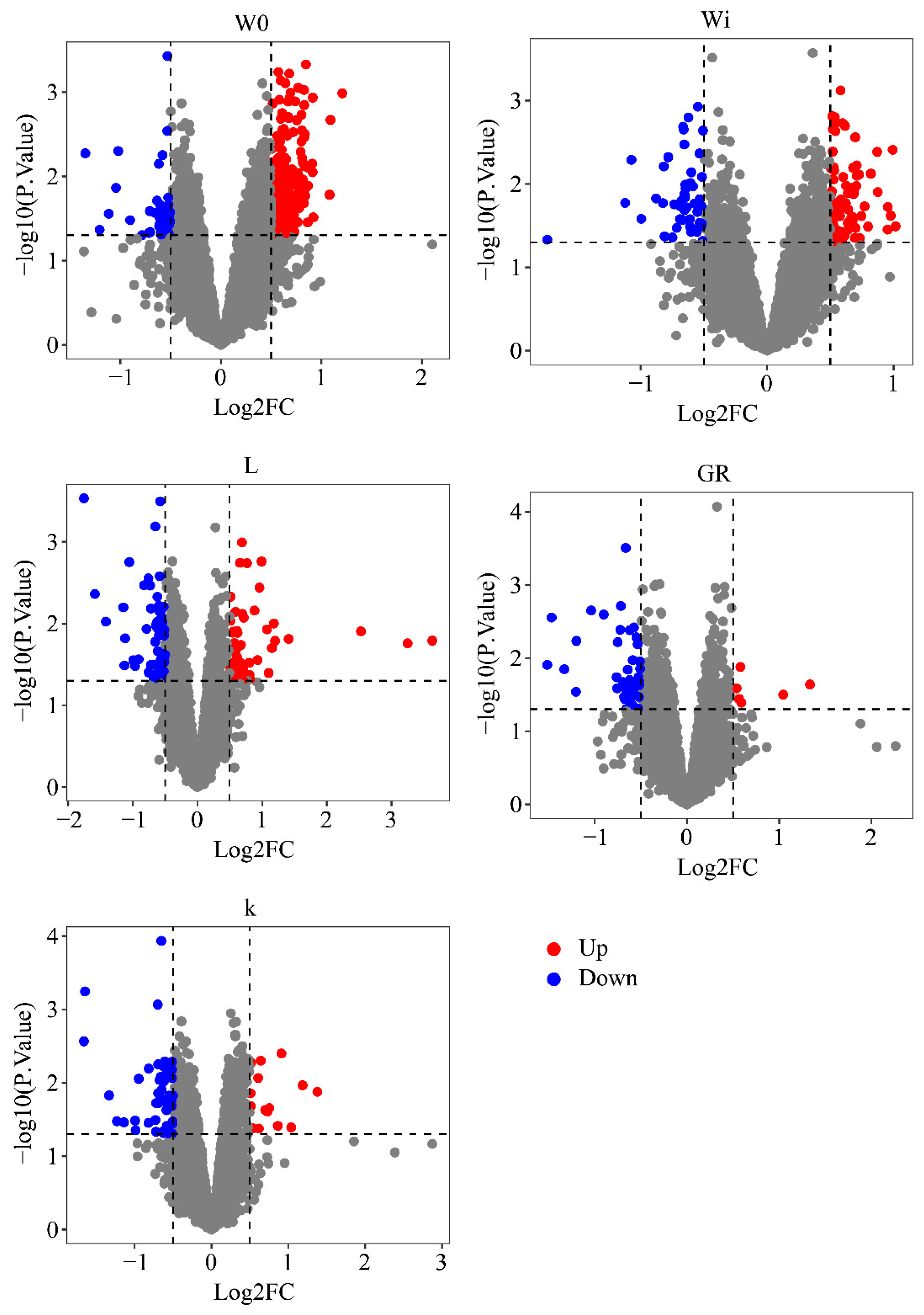
2.6. Differential Gene Expression Analysis
2.7. Genome-Wide Association Analysis
2.8. Gene Ontology Enrichment Analysis
2.9. Identification of Candidate Genes
3. Results
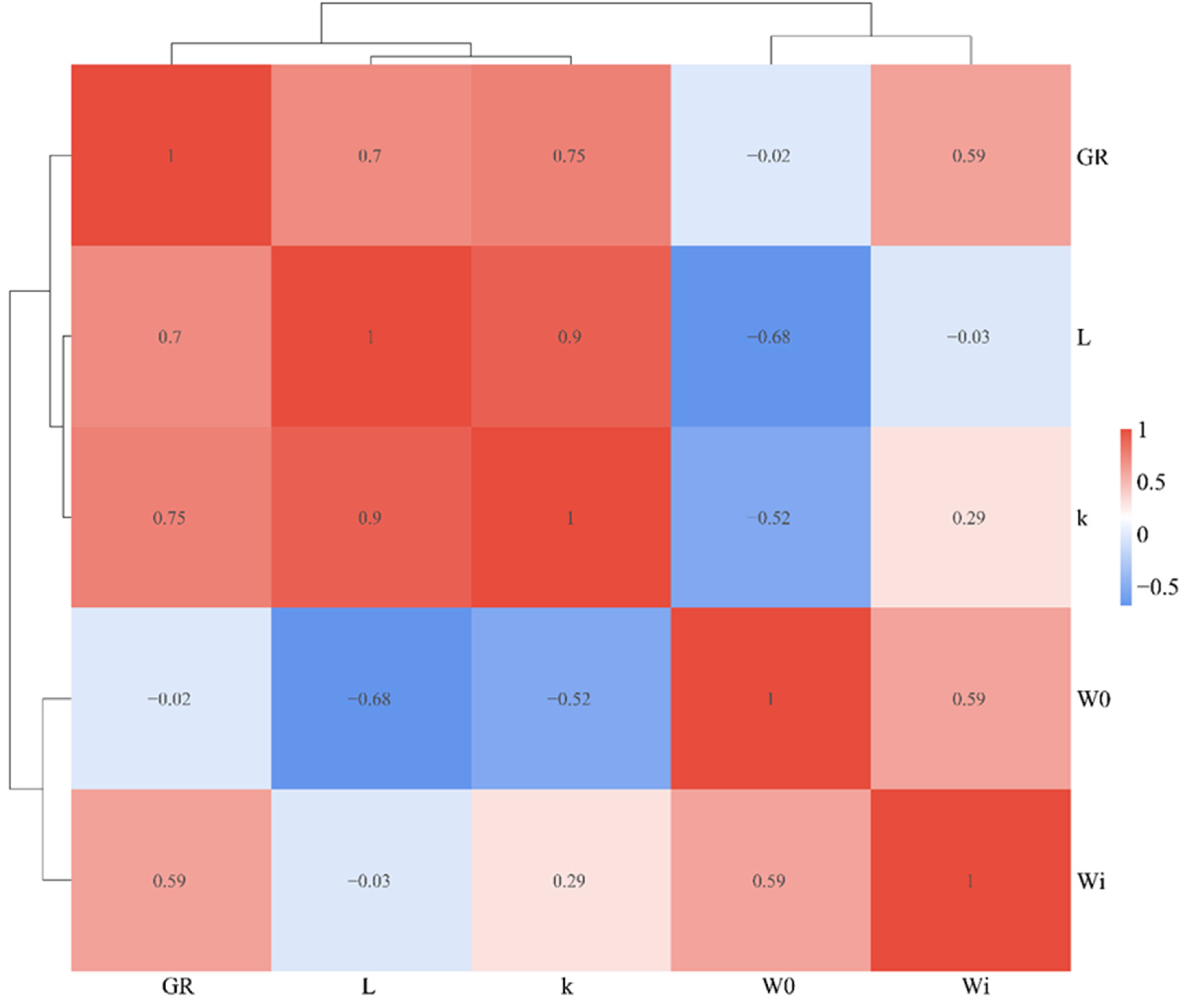
3.1. Growth Curve Fitting and Definition of Parameter Traits
3.2. Significant SNPs and Nearby Genes Identified by GWAS
3.3. WGCNA Candidate Genes and Differentially Expressed Genes Identified by Transcriptome Analysis
3.4. Integrated Analysis of Genome and Transcriptome Data and Gene Function Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Russo, V. A genomewide association study for average daily gain in Italian Large White pigs. J. Anim. Sci. 2014, 92, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.; Suzuki, K.; Kadowaki, H.; Shibata, T.; Oikawa, T. Genetic parameters for feed efficiency traits and their relationships with growth and carcass traits in Duroc pigs. J. Anim. Breed. Genet. 2007, 124, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zheng, Z.; Yu, H.; Wu, L.; Huang, H.; Jiang, P.; Li, X. Identification of factors affecting fattening efficiency of commercial pig herds and analysis of their impact at different performance levels. Sci. Rep. 2024, 14, 20105. [Google Scholar] [CrossRef] [PubMed]
- Dervishi, E.; Bai, X.; Dyck, M.K.; Harding, J.C.S.; Fortin, F.; Dekkers, J.C.M.; Plastow, G. GWAS and genetic and phenotypic correlations of plasma metabolites with complete blood count traits in healthy young pigs reveal implications for pig immune response. Front. Mol. Biosci. 2023, 10, 1140375. [Google Scholar] [CrossRef] [PubMed]
- Ibragimov, E.; Eriksen, E.; Nielsen, J.P.; Jørgensen, C.B.; Fredholm, M.; Karlskov-Mortensen, P. Towards identification of new genetic determinants for post-weaning diarrhea in piglets. Anim. Genet. 2024, 55, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Lunney, J.K.; Steibel, J.P.; Reecy, J.M.; Fritz, E.; Rothschild, M.F.; Kerrigan, M.; Trible, B.; Rowland, R.R. Probing genetic control of swine responses to PRRSV infection: Current progress of the PRRS host genetics consortium. BMC Proc. 2011, 5 (Suppl. S4), S30. [Google Scholar] [CrossRef]
- Tang, Z.; Xu, J.; Yin, L.; Yin, D.; Zhu, M.; Yu, M.; Li, X.; Zhao, S.; Liu, X. Genome-wide association study reveals candidate genes for growth relevant traits in pigs. Front. Genet. 2019, 10, 302. [Google Scholar] [CrossRef]
- Yang, X.; Deng, F.; Wu, Z.; Chen, S.-Y.; Shi, Y.; Jia, X.; Hu, S.; Wang, J.; Cao, W.; Lai, S.-J. A genome-wide association study identifying genetic variants associated with growth, carcass and meat quality traits in rabbits. Animals 2020, 10, 1068. [Google Scholar] [CrossRef]
- Forni, S.; Piles, M.; Blasco, A.; Varona, L.; Oliveira, H.N.D.; Lôbo, R.B.; Albuquerque, L.G.D. Comparison of different nonlinear functions to describe Nelore cattle growth. J. Anim. Sci. 2009, 87, 496–506. [Google Scholar] [CrossRef]
- Duan, X.; An, B.; Du, L.; Chang, T.; Liang, M.; Yang, B.-G.; Xu, L.; Zhang, L.; Li, J.; E, G.; et al. Genome-wide association analysis of growth curve parameters in Chinese Simmental beef cattle. Animals 2021, 11, 192. [Google Scholar] [CrossRef]
- Tjørve, K.M.; Tjørve, E. The use of Gompertz models in growth analyses, and new Gompertz-model approach: An addition to the Unified-Richards family. PLoS ONE 2017, 12, e0178691. [Google Scholar] [CrossRef] [PubMed]
- Aggrey, S.E. Comparison of three nonlinear and spline regression models for describing chicken growth curves. Poult. Sci. 2002, 81, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Starck, J.M.; Ricklefs, R.E. Avian Growth and Development: Evolution within the Altricial-Precocial Spectrum; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Seifi Moroudi, R.; Ansari Mahyari, S.; Vaez Torshizi, R.; Lanjanian, H.; Masoudi-Nejad, A.J.A.G. Identification of new genes and quantitative trait locis associated with growth curve parameters in F2 chicken population using genome-wide association study. Anim. Genet. 2021, 52, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Marinho, K.N.D.S.; Freitas, A.R.D.; Falcão, A.J.D.S.; Dias, F.E.F. Nonlinear models for fitting growth curves of Nellore cows reared in the Amazon Biome. Rev. Bras. Zootec. 2013, 42, 645–650. [Google Scholar] [CrossRef]
- Danil, K.; Chivers, S.J. Growth and reproduction of female short-beaked common dolphins, Delphinus delphis, in the eastern tropical Pacific. Can. J. Zool. 2007, 85, 108–121. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, Z.; Glória, L.S.; Zhang, K.; Zhang, C.; Yang, R.; Luo, X.; Jia, X.; Lai, S.-J.; Chen, S.-Y. Genome-wide association studies for growth curves in meat rabbits through the single-step nonlinear mixed model. Front. Genet. 2021, 12, 750939. [Google Scholar] [CrossRef]
- Silva, F.F.E.; Zambrano, M.F.B.; Varona, L.; Glória, L.S.; Lopes, P.S.; Silva, M.V.G.B.; Arbex, W.; Lázaro, S.F.; Resende, M.D.V.D.; Guimarães, S.E.F. Genome association study through nonlinear mixed models revealed new candidate genes for pig growth curves. Sci. Agric. 2017, 74, 1–7. [Google Scholar] [CrossRef]
- Ding, R.; Yang, M.; Wang, X.; Quan, J.; Zhuang, Z.; Zhou, S.; Li, S.; Xu, Z.; Zheng, E.; Cai, G.; et al. Genetic Architecture of Feeding Behavior and Feed Efficiency in a Duroc Pig Population. Front. Genet. 2018, 9, 220. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Meng, Q.; Wang, K.; Liu, X.; Zhou, H.; Xu, L.; Wang, Z.; Fang, M. Identification of growth trait related genes in a Yorkshire purebred pig population by genome-wide association studies. Asian-Australas. J. Anim. Sci. 2017, 30, 462–469. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Li, Y.Q.; Wu, H.Y.; Hu, B.; Zheng, J.J.; Zhai, H.; Lv, S.X.; Liu, X.L.; Chen, X.; Qiu, H.M.; et al. Genotyping of Soybean Cultivars With Medium-Density Array Reveals the Population Structure and QTNs Underlying Maturity and Seed Traits. Front. Plant Sci. 2018, 9, 610. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 2019, 8, giy154. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Lin, S.; Jiang, J.; Zeng, Y.; Meng, Y.; Ren, J.; Wu, P. Dual-Model GWAS Analysis and Genomic Selection of Maize Flowering Time-Related Traits. Genes 2024, 15, 740. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shi, H.; Yu, H.; Ma, Y.; Hu, H.; Han, Z.; Zhang, Y.; Zhen, Z.; Yi, L.; Hou, J. Combined GWAS and Transcriptome Analyses Provide New Insights Into the Response Mechanisms of Sunflower Against Drought Stress. Front. Plant Sci. 2022, 13, 847435. [Google Scholar] [CrossRef]
- Deng, T.; Liang, A.; Liang, S.; Ma, X.; Lu, X.; Duan, A.; Pang, C.; Hua, G.; Liu, S.; Campanile, G.; et al. Integrative Analysis of Transcriptome and GWAS Data to Identify the Hub Genes Associated With Milk Yield Trait in Buffalo. Front. Genet. 2019, 10, 36. [Google Scholar] [CrossRef]
- Liu, X.; Huang, J.; Yang, S.; Zhao, Y.; Xiang, A.; Cao, J.; Fan, B.; Wu, Z.; Zhao, J.; Zhao, S.; et al. Whole blood transcriptome comparison of pigs with extreme production of in vivo dsRNA-induced serum IFN-a. Dev. Comp. Immunol. 2014, 44, 35–43. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 25, 4–7. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Han, P.; Wang, C.; Zhang, W.; Wu, Y.; Wang, D.; Zhao, S.; Zhu, M. Pleiotropic architectures of porcine immune and growth trait pairs revealed by a self-product-based transcriptome method. Anim. Genet. 2023, 54, 123–131. [Google Scholar] [CrossRef]
- Gregory Alvord, W.; Roayaei, J.A.; Quiñones, O.A.; Schneider, K.T. A microarray analysis for differential gene expression in the soybean genome using Bioconductor and R. Brief. Bioinform. 2007, 8, 415–431. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Sun, F.; Zhao, Y.; Shan, L.; Lou, H. Identification of susceptibility modules and genes for cardiovascular disease in diabetic patients using WGCNA analysis. J. Diabetes Res. 2020, 2020, 4178639. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. GAPIT version 3: Boosting power and accuracy for genomic association and prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.J.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Rizzi, C.; Contiero, B.; Cassandro, M. Growth patterns of Italian local chicken populations. Poult. Sci. 2013, 92, 2226–2235. [Google Scholar] [CrossRef]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Conneely, K.N.; Boehnke, M. So many correlated tests, so little time! Rapid adjustment of P values for multiple correlated tests. Am. J. Hum. Genet. 2007, 81, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.; Toosi, A.; Wolc, A.; Garrick, D.; Dekkers, J. Application of whole-genome prediction methods for genome-wide association studies: A Bayesian approach. J. Agric. Biol. Environ. Stat. 2017, 22, 172–193. [Google Scholar] [CrossRef]
- Weller, J.I.; Shlezinger, M.; Ron, M. Correcting for bias in estimation of quantitative trait loci effects. Genet. Sel. Evol. 2005, 37, 501. [Google Scholar] [CrossRef]
- Misra, G.; Badoni, S.; Domingo, C.J.; Cuevas, R.P.O.; Llorente, C.; Mbanjo, E.G.N.; Sreenivasulu, N. Deciphering the genetic architecture of cooked rice texture. Front. Plant Sci. 2018, 9, 409256. [Google Scholar] [CrossRef]
- Yu, Z.; Chang, F.; Lv, W.; Sharmin, R.A.; Wang, Z.; Kong, J.; Bhat, J.A.; Zhao, T.J.G. Identification of QTN and candidate gene for seed-flooding tolerance in soybean [Glycine max (L.) Merr.] using genome-wide association study (GWAS). Genes 2019, 10, 957. [Google Scholar] [CrossRef]
- Gao, G.; Fiorica, P.N.; McClellan, J.; Barbeira, A.N.; Li, J.L.; Olopade, O.I.; Im, H.K.; Huo, D. A joint transcriptome-wide association study across multiple tissues identifies candidate breast cancer susceptibility genes. Am. J. Hum. Genet. 2023, 110, 950–962. [Google Scholar] [CrossRef]
- Page, R.; Huang, S.; Ronen, M.; Sela, H.; Sharon, A.; Shrestha, S.; Poland, J.; Steffenson, B.J. Genome-wide association mapping of rust resistance in Aegilops longissima. Front. Plant Sci. 2023, 14, 1196486. [Google Scholar] [CrossRef]
- Waiho, K.; Fazhan, H.; Shahreza, M.S.; Moh, J.H.Z.; Noorbaiduri, S.; Wong, L.L.; Sinnasamy, S.; Ikhwanuddin, M. Transcriptome Analysis and Differential Gene Expression on the Testis of Orange Mud Crab, Scylla olivacea, during Sexual Maturation. PLoS ONE 2017, 12, e0171095. [Google Scholar] [CrossRef]
- Fredriksson, L.; Li, H.; Eriksson, U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor. Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef]
- Shibuya, M. Structure and Function of VEGF/VEGF-receptor System Involved in Angiogenesis. Cell Struct. Funct. 2001, 26, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, A.; Alitalo, A.; Korpisalo, P.; Soronen, J.; Kaijalainen, S.; Leppanen, V.M.; Jeltsch, M.; Yla-Herttuala, S.; Alitalo, K. Activated forms of VEGF-C and VEGF-D provide improved vascular function in skeletal muscle. Circ. Res. 2009, 104, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Basciani, S.; Mariani, S.; Arizzi, M.; Ulisse, S.; Rucci, N.; Jannini, E.A.; Rocca, C.D.; Manicone, A.; Carani, C.; Spera, G.; et al. Expression of Platelet-Derived Growth Factor-A (PDGF-A), PDGF-B, and PDGF Receptor-α and -β during Human Testicular Development and Disease. J. Clin. Endocrinol. Metab. 2002, 87, 2310–2319. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.; Wu, D.; Matsumura, F.; Wei, Q. Centrosome/Spindle pole–associated protein regulates cytokinesis via promoting the recruitment of MyoGEF to the central spindle. Mol. Biol. Cell 2009, 20, 1428–1440. [Google Scholar] [CrossRef]
- Patzke, S.; Hauge, H.; Sioud, M.; Finne, E.F.; Sivertsen, E.A.; Delabie, J.; Stokke, T.; Aasheim, H.-C. Identification of a novel centrosome/microtubule-associated coiled-coil protein involved in cell-cycle progression and spindle organization. Oncogene 2005, 24, 1159–1173. [Google Scholar] [CrossRef]
- Patzke, S.; Redick, S.; Warsame, A.; Murga-Zamalloa, C.A.; Khanna, H.; Doxsey, S.; Stokke, T. CSPP is a ciliary protein interacting with Nephrocystin 8 and required for cilia formation. Mol. Biol. Cell 2010, 21, 2555–2567. [Google Scholar] [CrossRef]
- Patzke, S.; Stokke, T.; Aasheim, H.C. CSPP and CSPP-L associate with centrosomes and microtubules and differently affect microtubule organization. J. Cell. Physiol. 2006, 209, 199–210. [Google Scholar] [CrossRef]
- de Nijs, L.; Leon, C.; Nguyen, L.; Loturco, J.J.; Delgado-Escueta, A.V.; Grisar, T.; Lakaye, B. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat. Neurosci. 2009, 12, 1266–1274. [Google Scholar] [CrossRef]
- Kim, J.; Choi, B.; Lim, H.; Park, E.; Lee, S.; Seo, B.; Cho, I.; Lee, J.; Oh, S.; Jeon, J.T. Characterization of phosphoinositide-3-kinase, class 3 (PIK3C3) gene and association tests with quantitative traits in pigs. Asian-Australas. J. Anim. Sci. 2005, 18, 1701–1707. [Google Scholar] [CrossRef]
- Berndt, S.I.; Gustafsson, S.; Magi, R.; Ganna, A.; Wheeler, E.; Feitosa, M.F.; Justice, A.E.; Monda, K.L.; Croteau-Chonka, D.C.; Day, F.R.; et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat. Genet. 2013, 45, 501–512. [Google Scholar] [CrossRef]
- Jang, J.; Kim, B.; Jhang, S.Y.; Ahn, B.; Kang, M.; Park, C.; Cho, E.S.; Kim, Y.S.; Park, W.; Kim, H. Population differentiated copy number variation between Eurasian wild boar and domesticated pig populations. Sci. Rep. 2023, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, M.; Gao, P.; Cao, G.; Cheng, Z.; Zhang, W.; Liu, J.; Liu, X.; Li, B. Novel splice isoforms of pig myoneurin and their diverse mRNA expression patterns. Asian-Australas. J. Anim. Sci. 2018, 31, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ning, G.; Hou, Y.; Cao, Y.; Xu, J.; Wu, J.; Zhang, T.; Wang, Q. Myoneurin regulates BMP signaling by competing with Ppm1a for Smad binding. iScience 2022, 25, 104495. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Hildebrand, J.D.; Soriano, P. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol. Cell Biol. 2002, 22, 5296–5307. [Google Scholar] [CrossRef]
- Shen, L.; Li, Q.; Wang, J.; Zhao, Y.; Niu, L.; Bai, L.; Shuai, S.; Li, X.; Zhang, S.; Zhu, L. miR-144-3p Promotes Adipogenesis Through Releasing C/EBPalpha From Klf3 and CtBP2. Front. Genet. 2018, 9, 677. [Google Scholar] [CrossRef]
- Ponsuksili, S.; Murani, E.; Phatsara, C.; Schwerin, M.; Schellander, K.; Wimmers, K. Porcine muscle sensory attributes associate with major changes in gene networks involving CAPZB, ANKRD1, and CTBP2. Funct. Integr. Genom. 2009, 9, 455–471. [Google Scholar] [CrossRef]
- Jeong, H.; Choi, B.H.; Park, J.; Jung, J.H.; Shin, H.; Kang, K.W.; Quan, Y.H.; Yu, J.; Park, J.H.; Park, Y.; et al. GCC2 as a New Early Diagnostic Biomarker for Non-Small Cell Lung Cancer. Cancers 2021, 13, 5482. [Google Scholar] [CrossRef]
- Young, P.R. Perspective on the discovery and scientific impact of p38 MAP kinase. J. Biomol. Screen. 2013, 18, 1156–1163. [Google Scholar] [CrossRef]
- Gangwani, L. Deficiency of the Zinc Finger Protein ZPR1 Causes Defects in Transcription and Cell Cycle Progression. J. Biol. Chem. 2006, 281, 40330–40340. [Google Scholar] [CrossRef]
- Li, S.; Yu, W.; Hu, G. Angiogenin inhibits nuclear translocation of apoptosis inducing factor in a Bcl-2-dependent manner. J. Cell. Physiol. 2012, 227, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Tello-Montoliu, A.; Patel, J.; Lip, G.Y.H. Angiogenin: A review of the pathophysiology and potential clinical applications. J. Thromb. Haemost. 2006, 4, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-p.; Tsuji, T.; Riordan, J.F.; Hu, G.-F. Identification and characterization of an angiogenin-binding DNA sequence that stimulates luciferase reporter gene expression. Biochemistry 2003, 42, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Sooryanarain, H.; Rogers, A.J.; Cao, D.; Haac, M.E.R.; Karpe, Y.A.; Meng, X.-J. ISG15 modulates type I interferon signaling and the antiviral response during hepatitis E virus replication. J. Virol. 2017, 91, e02185-18. [Google Scholar] [CrossRef]
- Speer, S.D.; Li, Z.; Buta, S.; Payelle-Brogard, B.; Qian, L.; Vigant, F.; Rubino, E.; Gardner, T.J.; Wedeking, T.; Hermann, M. ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 2016, 7, 11496. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Hou, L.; Wu, W.; Zhao, S.; Xiong, Y. Fibroblast Growth Factor 21 Suppresses Adipogenesis in Pig Intramuscular Fat Cells. Int. J. Mol. Sci. 2015, 17, 11. [Google Scholar] [CrossRef]
- Cui, W.; Igata, T.; Tanaka, H.; Etoh, K.; Hong, S.; Tani, N.; Koga, T.; Nakao, M. Loss of the transcription repressor ZHX3 induces senescence-associated gene expression and mitochondrial-nucleolar activation. PLoS ONE 2022, 17, e0262488. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, D.; Ji, C. Zinc fingers and homeoboxes family in human diseases. Cancer Gene Ther. 2015, 22, 223–226. [Google Scholar] [CrossRef]
- Suehiro, F.; Nishimura, M.; Kawamoto, T.; Kanawa, M.; Yoshizawa, Y.; Murata, H.; Kato, Y. Impact of zinc fingers and homeoboxes 3 on the regulation of mesenchymal stem cell osteogenic differentiation. Stem Cells Dev. 2011, 20, 1539–1547. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, S.; Zhou, H.; Cao, D. Low expression of ZHX3 is associated with progression and poor prognosis in colorectal cancer. Transl. Oncol. 2024, 39, 101829. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gompertz–Laird | Functions | |
|---|---|---|
| Model | 0.9952 ± 0.0066/0.9950 ± 0.0068 | |
| Parameters | ||
| Base parameter | ||
| Base parameter | ||
| Base parameter | ||
| Traits | Mean | SD | Minimum | Maximum | |
|---|---|---|---|---|---|
| 1.4690 | 0.3452 | 0.5214 | 2.3429 | 0.4183 ± 0.0004 | |
| L | 0.0500 | 0.0209 | 0.0149 | 0.1683 | 0.4152 ± 0.0004 |
| k | 0.0100 | 0.0109 | 0.0176 | 0.0716 | 0.4865 ± 0.0005 |
| 1.7196 | 0.4273 | 0.3716 | 3.0714 | 0.3101 ± 0.0004 | |
| GR | 0.0692 | 0.0196 | 0.0181 | 0.1393 | 0.3600 ± 0.0004 |
| SNP | Traits | Chromosome | Position | FarmCPU | BLINK | Near Genes |
|---|---|---|---|---|---|---|
| INRA0056566 | X | 12085635 | *** | *** | ACE2 | |
| AP1S2 | ||||||
| ASB9 | ||||||
| ASB11 | ||||||
| BMX | ||||||
| CLTRN | ||||||
| PIGA | ||||||
| PIR | ||||||
| VEGFD | ||||||
| ZRSR2 | ||||||
| DRGA0004151 | 3 | 48199203 | ** | * | GCC2 | |
| SLC5A7 | ||||||
| ST6GAL2 | ||||||
| SULT1C2 | ||||||
| SULT1C3 | ||||||
| SULT1C4 | ||||||
| U6 | ||||||
| INRA0056460 | X | 6388935 | ** | *** | SHROOM2 | |
| U6 | ||||||
| WWC3 | ||||||
| H3GA0049324 | 17 | 44259688 | *** | *** | CHD6 | |
| *** | *** | EMILIN3 | ||||
| LPIN3 | ||||||
| PLCG1 | ||||||
| PTPRT | ||||||
| ZHX3 | ||||||
| H3GA0037747 | 13 | 108017340 | * | * | ACTRT3 | |
| LRRC34 | ||||||
| LRRIQ4 | ||||||
| MECOM | ||||||
| MYNN |
| Traits | Candidate Genes | Term Ratio | Source |
|---|---|---|---|
| VEGFD | 4/5 | SGGs | |
| PDGFA | 4/5 | DEGs | |
| CSPP1 | 3/5 | DEGs | |
| EFHC1 | 2/5 | DEGs | |
| PIK3C3 | 2/5 | DEGs | |
| ZZZ3 | 2/5 | DEGs | |
| GCC2 | 9/12 | SGGs | |
| MAPK14 | 9/12 | DEGs | |
| ZPR1 | 7/12 | WCGs | |
| ISG15 | 5/12 | DEGs | |
| ANG | 4/12 | DEGs | |
| ZHX3 | 2/2 | SGGs | |
| CEBPD | 2/2 | DEGs | |
| MYNN | 2/2 | SGGs | |
| CTBP2 | 2/2 | WCGs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, Z.; Qiao, J.; Xu, F.; Li, X.; Zhao, Y.; Zhu, M. Integrated Analysis Reveals Genetic Basis of Growth Curve Parameters in an F2 Designed Pig Population Based on Genome and Transcriptome Data. Agriculture 2024, 14, 1704. https://doi.org/10.3390/agriculture14101704
Che Z, Qiao J, Xu F, Li X, Zhao Y, Zhu M. Integrated Analysis Reveals Genetic Basis of Growth Curve Parameters in an F2 Designed Pig Population Based on Genome and Transcriptome Data. Agriculture. 2024; 14(10):1704. https://doi.org/10.3390/agriculture14101704
Chicago/Turabian StyleChe, Zhaoxuan, Jiakun Qiao, Fangjun Xu, Xinyun Li, Yunxia Zhao, and Mengjin Zhu. 2024. "Integrated Analysis Reveals Genetic Basis of Growth Curve Parameters in an F2 Designed Pig Population Based on Genome and Transcriptome Data" Agriculture 14, no. 10: 1704. https://doi.org/10.3390/agriculture14101704
APA StyleChe, Z., Qiao, J., Xu, F., Li, X., Zhao, Y., & Zhu, M. (2024). Integrated Analysis Reveals Genetic Basis of Growth Curve Parameters in an F2 Designed Pig Population Based on Genome and Transcriptome Data. Agriculture, 14(10), 1704. https://doi.org/10.3390/agriculture14101704