
Comparative Transcriptome Analysis Provides Insights into Fruit Trichome Development in Peach




Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Phenotypic Analysis
2.2. Total RNA Extraction
2.3. Library Preparation and Sequencing
2.4. Transcriptome Sequencing Data Analysis
2.5. Candidate Genes for Trichome Development
2.6. The Promoter Analysis of Candidate Genes
2.7. Quantitative Real-Time PCR Analysis
3. Results
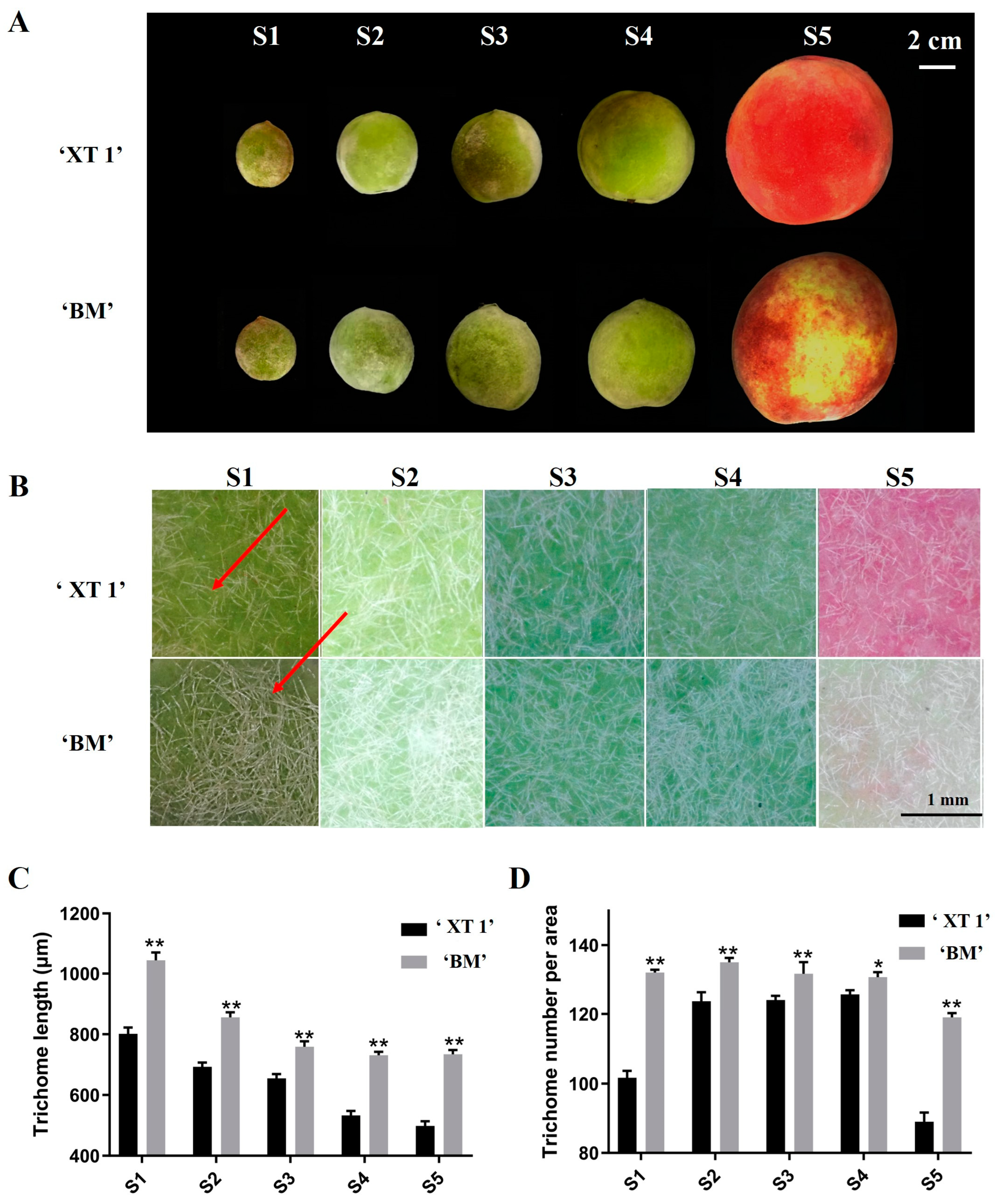
3.1. Phenotypic Differences in Fruit Trichome between ‘XT1’ and ‘BM’ Peaches
3.2. Overview of Transcriptome Sequencing Data
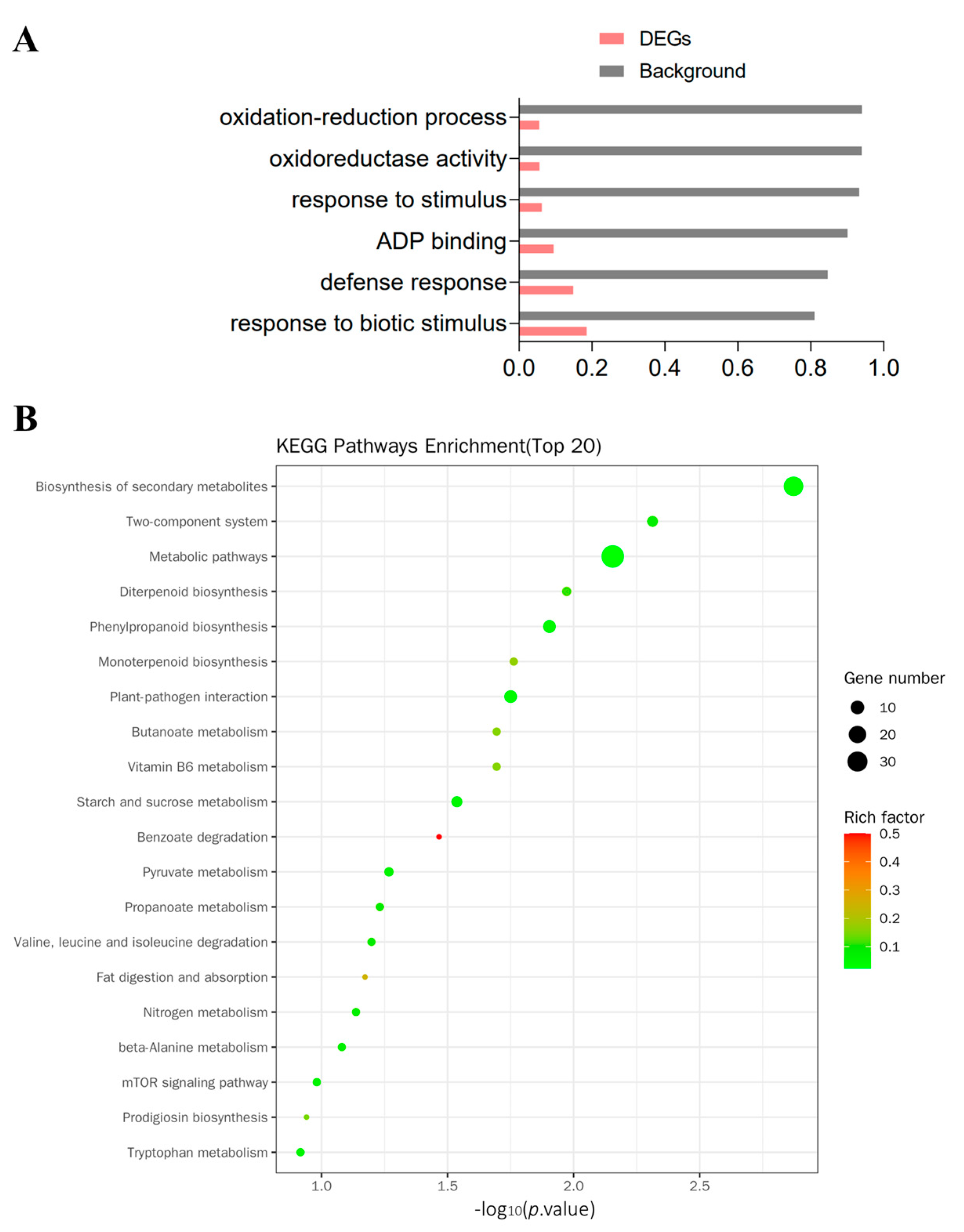
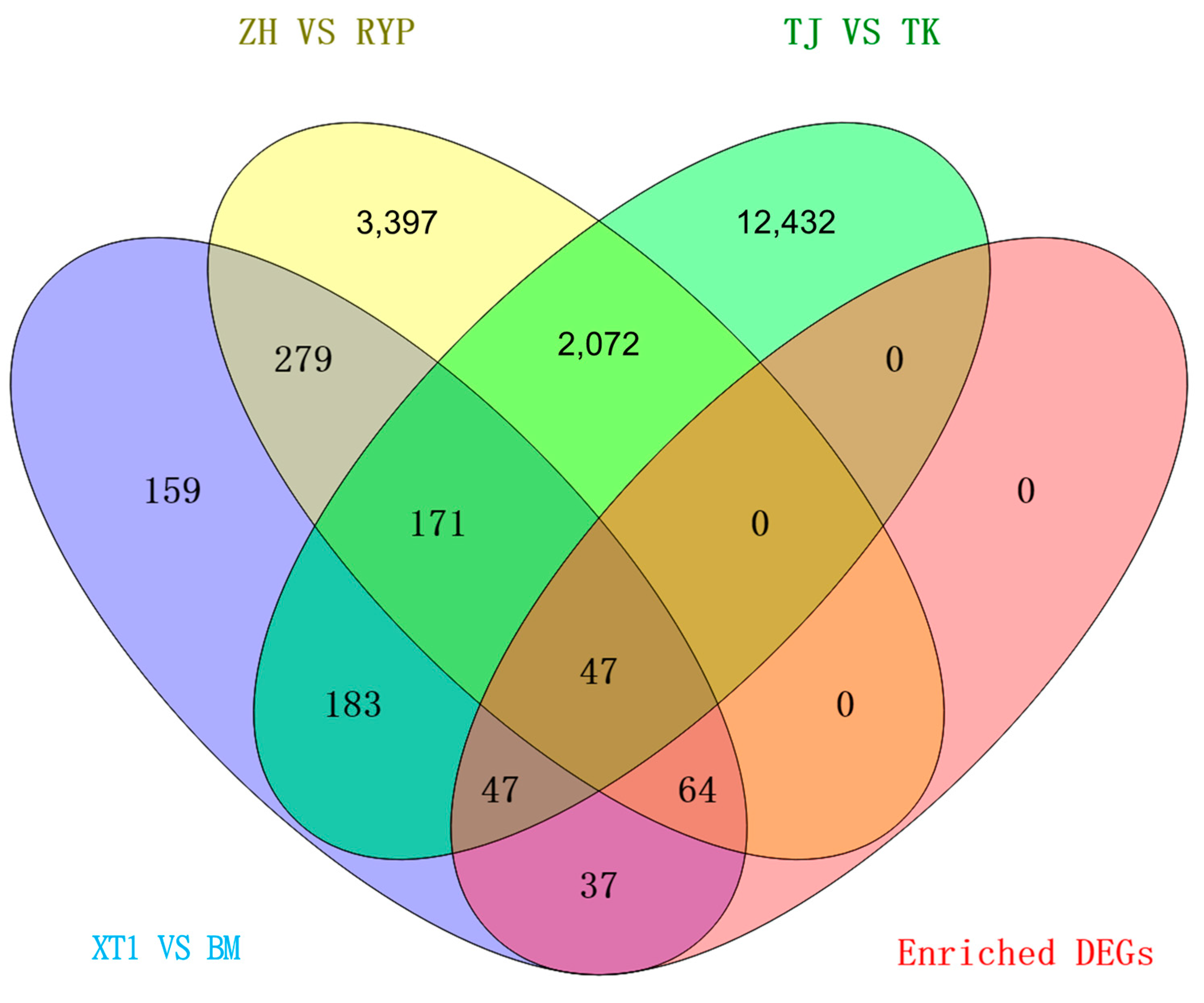
3.3. Functional Annotation of DEGs
3.4. Selection of Genes for Trichome Development
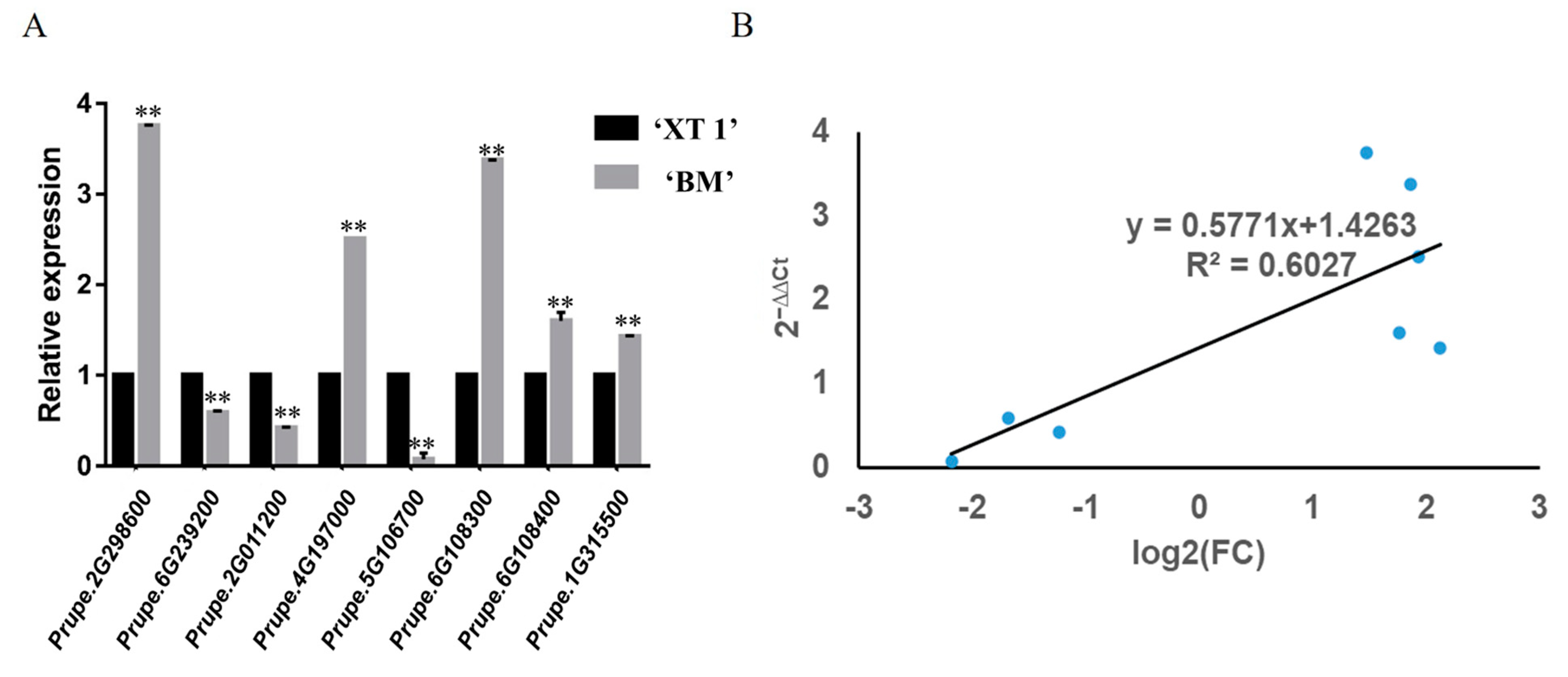
3.5. qRT-PCR Verification of DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Q.; Yang, X.; Wang, L.; Zheng, B.; Cai, Y.; Ogutu, C.O.; Zhao, L.; Peng, Q.; Liao, L.; Zhao, Y.; et al. Two R2R3-MYB genes cooperatively control trichome development and cuticular wax biosynthesis in Prunus persica. New Phytol. 2022, 234, 179–196. [Google Scholar] [CrossRef]
- Han, G.; Li, Y.; Yang, Z.; Wang, C.; Zhang, Y.; Wang, B. Molecular Mechanisms of Plant Trichome Development. Front. Plant Sci. 2022, 13, 910228. [Google Scholar] [CrossRef] [PubMed]
- Hulskamp, M. Trichomes. Curr. Biol. 2019, 29, R273–R274. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, V.; Khayet, M.; Montero-Prado, P.; Heredia-Guerrero, J.A.; Liakopoulos, G.; Karabourniotis, G.; Del Rio, V.; Dominguez, E.; Tacchini, I.; Nerin, C.; et al. New insights into the properties of pubescent surfaces: Peach fruit as a model. Plant Physiol. 2011, 156, 2098–2108. [Google Scholar] [CrossRef]
- Creller, M.A.; Werner, D.J. Characterizing the novel fruit surface morphology of ‘Marina’ peach using scanning electron microscopy. J. Am. Soc. Hortic. Sci. 1996, 121, 198–203. [Google Scholar] [CrossRef]
- Gao, Z.S.; Zhou, X.; Yang, Z.W.; Versteeg, S.A.; Gao, L.; Fu, W.Y.; Wang, H.Y.; Zhou, J.Y.; Akkerdaas, J.H.; van Ree, R. IgE-binding potencies of three peach Pru p 1 isoforms. Mol. Nutr. Food Res. 2016, 60, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Song, S.S.; Liu, B.; Song, J.Q.; Pang, S.A.; Song, T.X.; Gao, S.; Zhang, Y.; Huang, H.; Qi, T.C. A molecular framework for signaling crosstalk between jasmonate and ethylene in anthocyanin biosynthesis, trichome development, and defenses against insect herbivores in Arabidopsis. J. Integr. Plant Biol. 2022, 64, 1770–1788. [Google Scholar] [CrossRef]
- Vendramin, E.; Pea, G.; Dondini, L.; Pacheco, I.; Dettori, M.T.; Gazza, L.; Scalabrin, S.; Strozzi, F.; Tartarini, S.; Bassi, D.; et al. A unique mutation in a MYB gene cosegregates with the nectarine phenotype in peach. PLoS ONE 2014, 9, e90574. [Google Scholar] [CrossRef]
- Khosla, A.; Paper, J.M.; Boehler, A.P.; Bradley, A.M.; Neumann, T.R.; Schrick, K. HD-Zip Proteins GL2 and HDG11 Have Redundant Functions in Arabidopsis Trichomes, and GL2 Activates a Positive Feedback Loop via MYB23. Plant Cell 2014, 26, 2184–2200. [Google Scholar] [CrossRef]
- Shan, C.M.; Shangguan, X.X.; Zhao, B.; Zhang, X.F.; Chao, L.M.; Yang, C.Q.; Wang, L.J.; Zhu, H.Y.; Zeng, Y.D.; Guo, W.Z.; et al. Control of cotton fibre elongation by a homeodomain transcription factor GhHOX3. Nat. Commun. 2014, 5, 5519. [Google Scholar] [CrossRef]
- Lu, Z.; Pan, L.; Wei, B.; Niu, L.; Cui, G.; Wang, L.; Zeng, W.; Wang, Z. Fine Mapping of the Gene Controlling the Fruit Skin Hairiness of Prunus persica and Its Uses for MAS in Progenies. Plants 2021, 10, 1433. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, L.; Han, Y.; Ren, X.; Huang, J.; Chen, H. De novo transcriptome sequencing and analysis of Coccinella septempunctata L. in non-diapause, diapause and diapause-terminated states to identify diapause-associated genes. BMC Genom. 2015, 16, 1086. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, S.; Khan, A.R.; Gan, Y. TOE1/TOE2 Interacting with GIS to Control Trichome Development in Arabidopsis. Int. J. Mol. Sci. 2023, 24, 6698. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Cao, K.; Qin, A.; Liu, Z.; Guan, L.; Sun, S.; Liu, H.; Zhou, Y.; Yang, J.; Liu, Y.; et al. Comparative spatial transcriptomics of peach and nectarine fruits elucidates the mechanism underlying fruit trichome development. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hua, B.; Chang, J.; Wu, M.L.; Xu, Z.J.; Zhang, F.Y.; Yang, M.N.; Xu, H.M.; Wang, L.J.; Chen, X.Y.; Wu, S. Mediation of JA signalling in glandular trichomes by thewoolly/SlMYC1regulatory module improves pest resistance in tomato. Plant Biotechnol. J. 2021, 19, 375–393. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Niu, D.; Wang, Z.; Yan, X.; Yang, X.; Yang, Y.; Cui, H. Tobacco transcription repressors NtJAZ: Potential involvement in abiotic stress response and glandular trichome induction. Plant Physiol. Biochem. 2019, 141, 388–397. [Google Scholar] [CrossRef]
- Li, S.; Tosens, T.; Harley, P.C.; Jiang, Y.F.; Kanagendran, A.; Grosberg, M.; Jaamets, K.; Niinemets, U. Glandular trichomes as a barrier against atmospheric oxidative stress: Relationships with ozone uptake, leaf damage, and emission of LOX products across a diverse set of species. Plant Cell Environ. 2018, 41, 1263–1277. [Google Scholar] [CrossRef]
- Escobar-Bravo, R.; Ruijgrok, J.; Kim, H.K.; Grosser, K.; Van Dam, N.M.; Klinkhamer, P.G.L.; Leiss, K.A. Light Intensity-Mediated Induction of Trichome-Associated Allelochemicals Increases Resistance Against Thrips in Tomato. Plant Cell Physiol. 2018, 59, 2462–2475. [Google Scholar] [CrossRef] [PubMed]
- Zandalinas, S.I.; Balfagon, D.; Gomez-Cadenas, A.; Mittler, R. Responses of plants to climate change: Metabolic changes during abiotic stress combination in plants. J. Exp. Bot. 2022, 73, 3339–3354. [Google Scholar] [CrossRef]
- Sallets, A.; Beyaert, M.; Boutry, M.; Champagne, A. Comparative proteomics of short and tall glandular trichomes of Nicotiana tabacum reveals differential metabolic activities. J. Proteome Res. 2014, 13, 3386–3396. [Google Scholar] [CrossRef]
- Gao, S.; Li, N.; Niran, J.; Wang, F.; Yin, Y.; Yu, C.; Jiao, C.; Yang, C.; Yao, M. Transcriptome profiling of Capsicum annuum using Illumina- and PacBio SMRT-based RNA-Seq for in-depth understanding of genes involved in trichome formation. Sci. Rep. 2021, 11, 10164. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, H.X.; Zhou, D.D.; Li, C.; Ding, Q.; Yang, X.G.; Wang, F.D.; Zheng, H.; Gao, J.W. Genetic and Transcriptome Analysis of Leaf Trichome Development in Chinese Cabbage (Brassica rapa L. subsp. pekinensis) and Molecular Marker Development. Int. J. Mol. Sci. 2022, 23, 12721. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, F.; Tang, Y.; Yuan, Y.; Deng, W.; Li, Z. Auxin Response Gene SlARF3 Plays Multiple Roles in Tomato Development and is Involved in the Formation of Epidermal Cells and Trichomes. Plant Cell Physiol. 2015, 56, 2110–2124. [Google Scholar] [CrossRef]
- Zhang, X.; Cao, J.; Huang, C.; Zheng, Z.; Liu, X.; Shangguan, X.; Wang, L.; Zhang, Y.; Chen, Z. Characterization of cotton ARF factors and the role of GhARF2b in fiber development. BMC Genom. 2021, 22, 202. [Google Scholar] [CrossRef]
- Zhang, M.; Zheng, X.; Song, S.; Zeng, Q.; Hou, L.; Li, D.; Zhao, J.; Wei, Y.; Li, X.; Luo, M.; et al. Spatiotemporal manipulation of auxin biosynthesis in cotton ovule epidermal cells enhances fiber yield and quality. Nat. Biotechnol. 2011, 29, 453–458. [Google Scholar] [CrossRef]
- Liu, S.; Fan, L.; Liu, Z.; Yang, X.; Zhang, Z.; Duan, Z.; Liang, Q.; Imran, M.; Zhang, M.; Tian, Z. A Pd1-Ps-P1 Feedback Loop Controls Pubescence Density in Soybean. Mol. Plant 2020, 13, 1768–1783. [Google Scholar] [CrossRef]
- Worthen, J.M.; Yamburenko, M.V.; Lim, J.; Nimchuk, Z.L.; Kieber, J.J.; Schaller, G.E. Type-B response regulators of rice play key roles in growth, development and cytokinin signaling. Development 2019, 146, dev174870. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Wang, L.M.; Han, L.J.; Cheng, Z.H.; Liu, X.F.; Wang, S.Y.; Liu, L.; Chen, J.C.; Song, W.Y.; Zhao, J.Y.; et al. HECATE2 acts with GLABROUS3 and Tu to boost cytokinin biosynthesis and regulate cucumber fruit wart formation. Plant Physiol. 2021, 187, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, L.; Zhao, Y.; An, L.; Yan, A.; Meng, X.; Gan, Y. Zinc Finger Protein 6 (ZFP6) regulates trichome initiation by integrating gibberellin and cytokinin signaling in Arabidopsis thaliana. New Phytol. 2013, 198, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Liu, C.; Yu, H.; Broun, P. Integration of cytokinin and gibberellin signalling by Arabidopsis transcription factors GIS, ZFP8 and GIS2 in the regulation of epidermal cell fate. Development 2007, 134, 2073–2081. [Google Scholar] [CrossRef]
- Traw, M.B.; Bergelson, J. Interactive effects of jasmonic acid, salicylic acid, and gibberellin on induction of trichomes in Arabidopsis. Plant Physiol. 2003, 133, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Zhang, N.; Wang, W.; Ahmed, S.; Cheng, Y.; Chen, S.; Wang, X.; Wang, Y.; Hu, X.; Wang, T.; et al. Involvement of ABA Responsive SVB Genes in the Regulation of Trichome Formation in Arabidopsis. Int. J. Mol. Sci. 2021, 22, 6790. [Google Scholar] [CrossRef]
- Zhu, G.; Liu, Y.; Ye, N.; Liu, R.; Zhang, J. Involvement of the abscisic acid catabolic gene CYP707A2 in the glucose-induced delay in seed germination and post-germination growth of Arabidopsis. Physiol. Plant 2011, 143, 375–384. [Google Scholar] [CrossRef]
- Cai, Y.; Ma, Z.; Ogutu, C.O.; Zhao, L.; Liao, L.; Zheng, B.; Zhang, R.; Wang, L.; Han, Y. Potential Association of Reactive Oxygen Species With Male Sterility in Peach. Front. Plant Sci. 2021, 12, 653256. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Error (%) | Q20 (%) | Q30 (%) | GC (%) | Total Mapped |
|---|---|---|---|---|---|---|---|
| ‘XT1’-1 | 57,601,296 | 57,601,172 | 0.02 | 98.08 | 94.13 | 44.56 | 55,052,373 (95.58%) |
| ‘XT1’-2 | 74,360,894 | 74,360,768 | 0.02 | 97.98 | 93.86 | 44.58 | 70,067,482 (94.23%) |
| ‘XT1’-3 | 98,645,922 | 98,645,650 | 0.02 | 98.11 | 94.20 | 44.24 | 94,237,865 (95.53%) |
| ‘BM’-1 | 58,302,810 | 58,302,758 | 0.02 | 97.79 | 93.34 | 44.75 | 54,110,721 (92.81%) |
| ‘BM’-2 | 58,911,454 | 58,911,394 | 0.02 | 98.00 | 93.88 | 44.74 | 54,695,889 (92.84%) |
| ‘BM’-3 | 97,403,128 | 97,403,026 | 0.02 | 97.66 | 93.15 | 44.96 | 92,143,059 (94.60%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Xu, M.; Guo, J.; Gan, Y. Comparative Transcriptome Analysis Provides Insights into Fruit Trichome Development in Peach. Agriculture 2024, 14, 427. https://doi.org/10.3390/agriculture14030427
Liu Y, Xu M, Guo J, Gan Y. Comparative Transcriptome Analysis Provides Insights into Fruit Trichome Development in Peach. Agriculture. 2024; 14(3):427. https://doi.org/10.3390/agriculture14030427
Chicago/Turabian StyleLiu, Yihua, Meng Xu, Jian Guo, and Yinbo Gan. 2024. "Comparative Transcriptome Analysis Provides Insights into Fruit Trichome Development in Peach" Agriculture 14, no. 3: 427. https://doi.org/10.3390/agriculture14030427
APA StyleLiu, Y., Xu, M., Guo, J., & Gan, Y. (2024). Comparative Transcriptome Analysis Provides Insights into Fruit Trichome Development in Peach. Agriculture, 14(3), 427. https://doi.org/10.3390/agriculture14030427