
Mapping Novel Loci and Candidate Genes Associated with Cadmium Content in Maize Using Genome-Wide Association Analysis

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Phenotypic Data Analysis
2.2. Genome-Wide Association Mapping, LD Analysis, and Superior Allele Analysis
2.3. Candidate Gene Prediction, GO, and KEGG Analysis, and Bioinformatics Analysis
2.4. RT-qPCR and Yeast Cadmium Tolerance Experiment
3. Results
3.1. Statistical Analysis of Phenotype
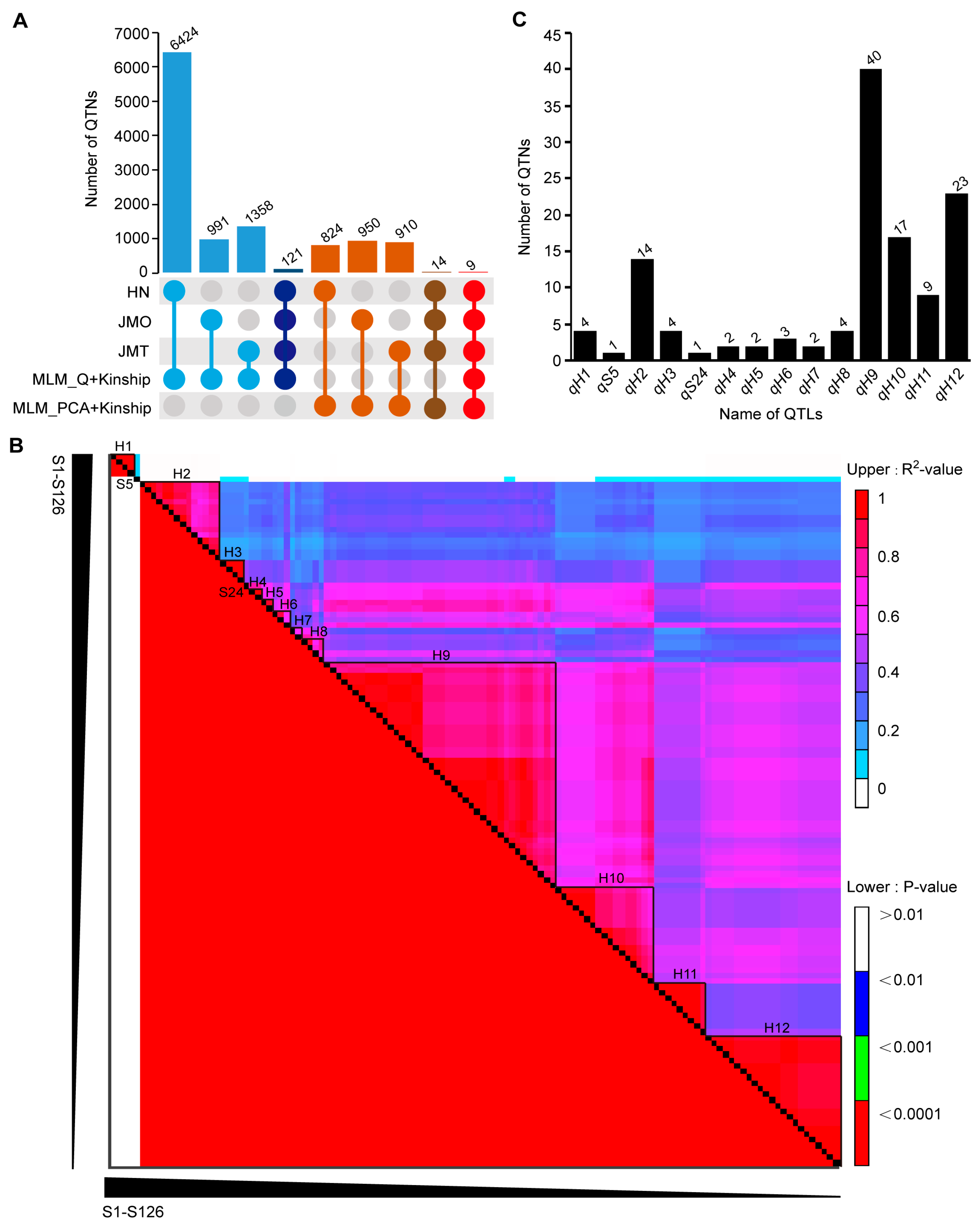
3.2. Genome-Wide Association Studies and Linkage Disequilibrium
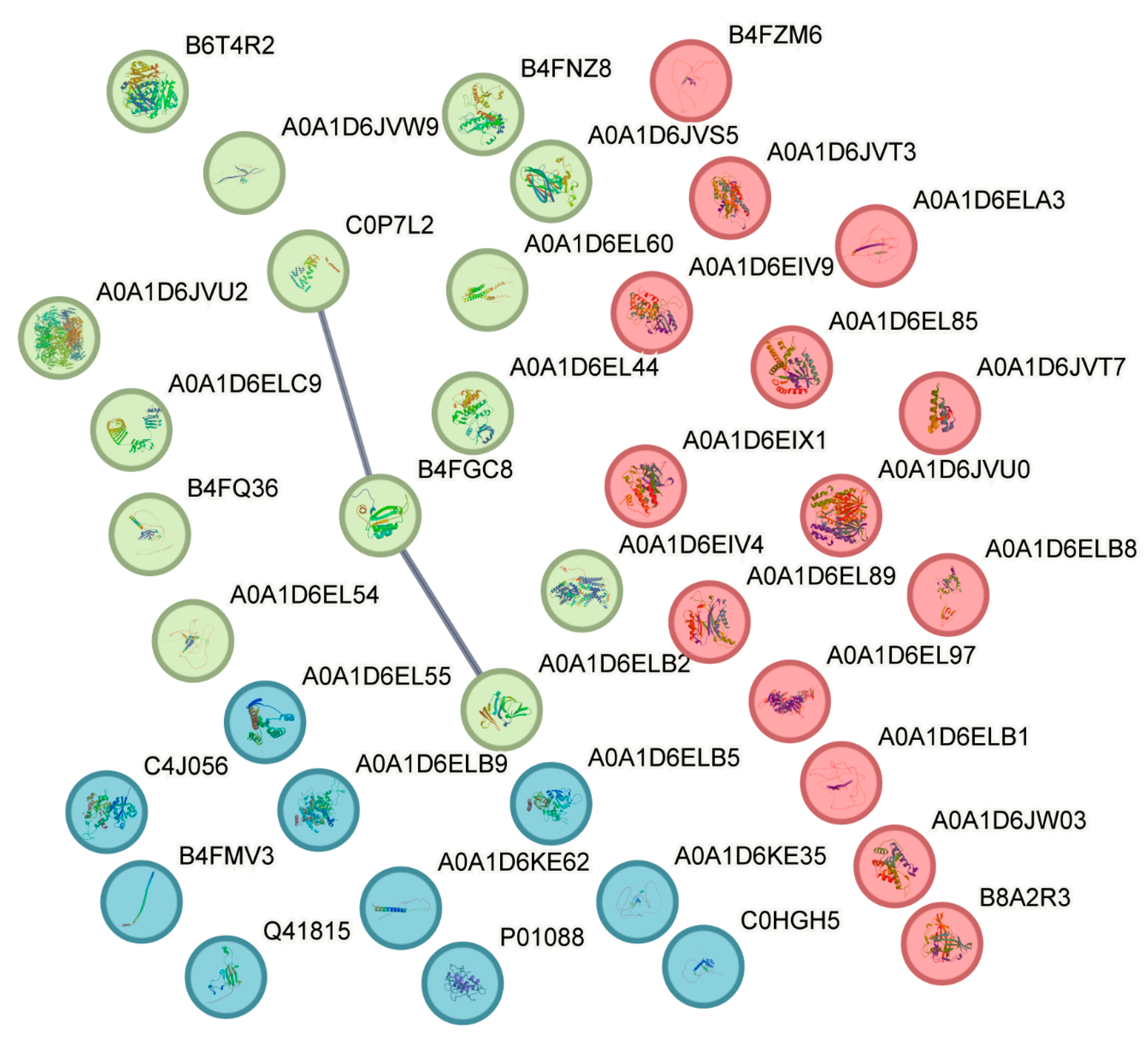
3.3. Candidate Gene Screening, Go and KEGG Analysis, and Interaction Analysis
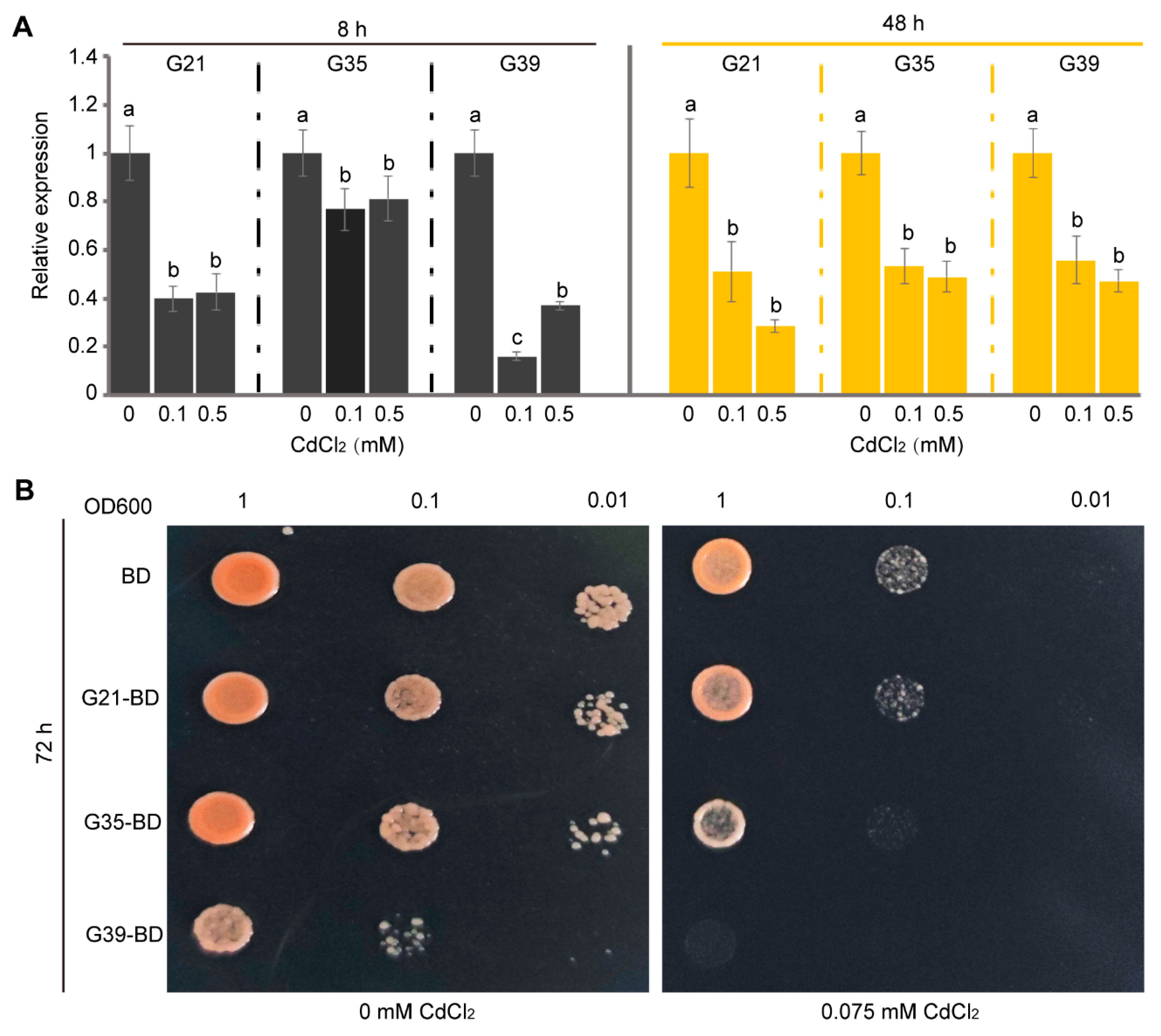
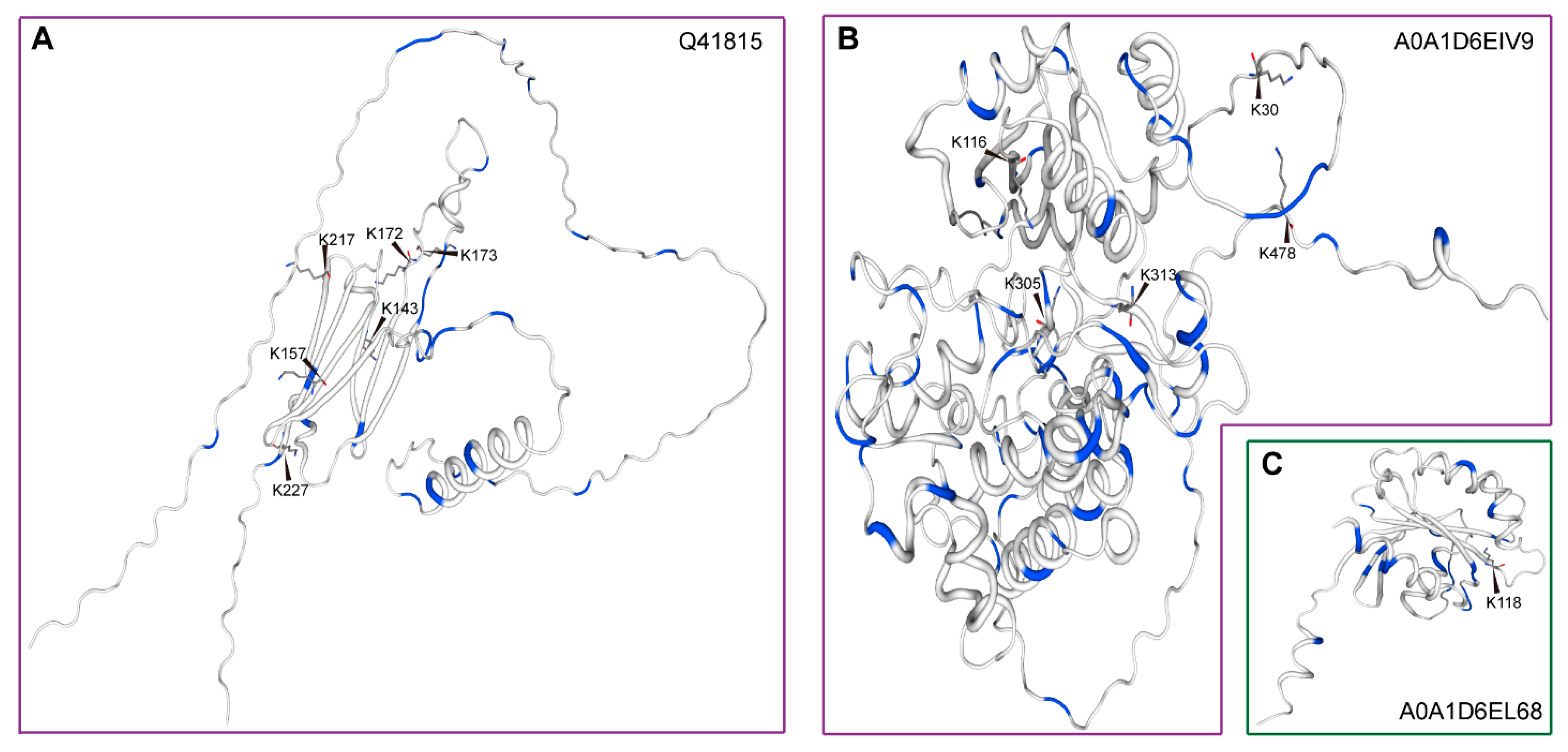
3.4. Gene Expression Profiles, Yeast Cadmium Tolerance, and Post-Translational Modifications Prediction
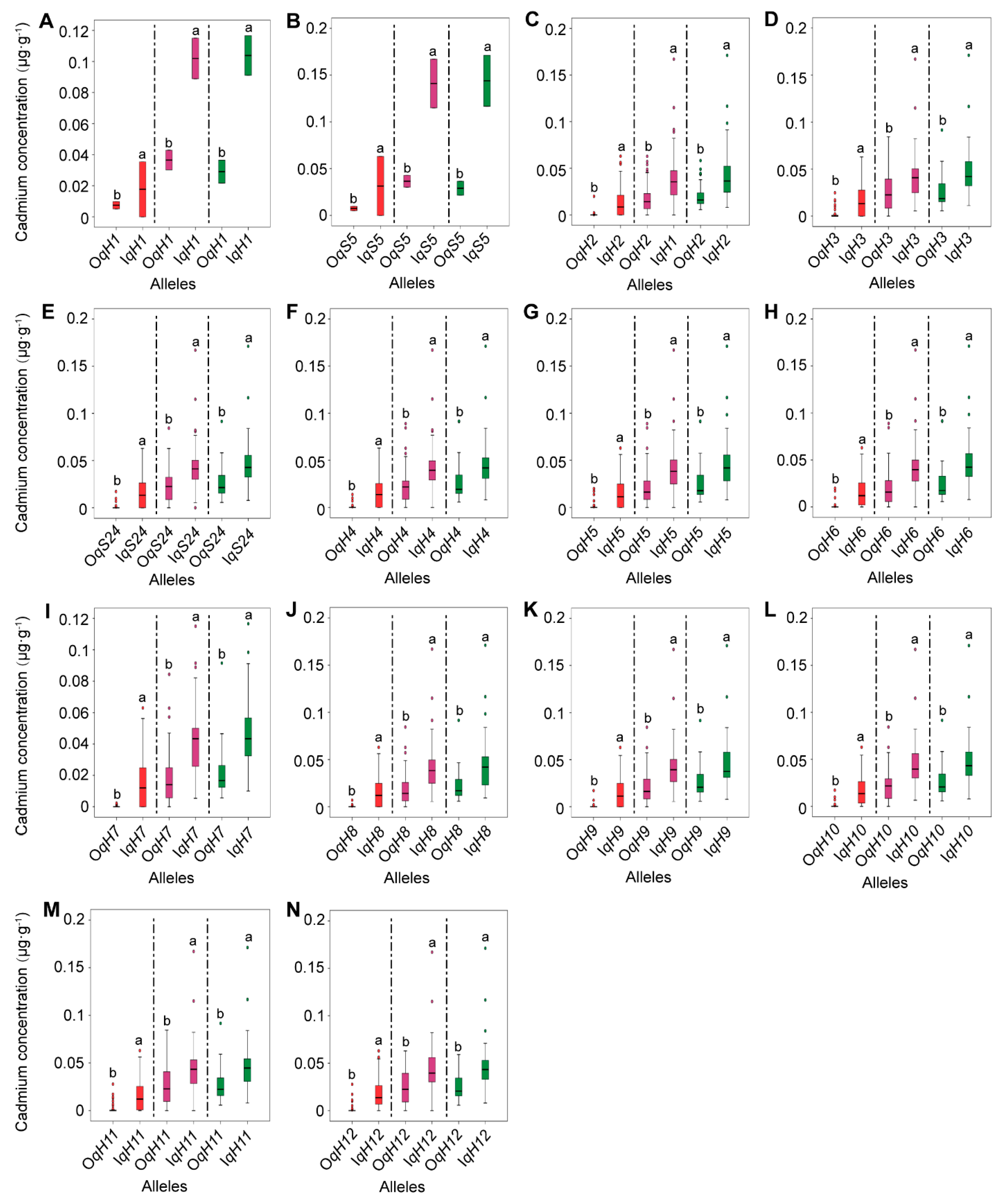
3.5. Distribution of Superior Alleles and Hybrid Combination Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Suleman, M.; Ashraf, M.; Raza, Q.U.A.; Bashir, M.A.; Rahman, S.U.; Aon, M.; Ali, S.; Shahzad, S.M.; Khalid, M.U.; Raza, H.M.A.; et al. Determining the Cadmium Accumulation in Maize (Zea mays L.) and Soil Influenced by Phosphoric Fertilizers in Two Different Textured Soils. Land 2022, 11, 1313. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, S.U.; Jung, K.Y.; Lee, S.; Choi, Y.D.; Owens, V.N.; Kumar, S.; Yun, S.W.; Hong, C.O. Cadmium Phytoavailability from 1976 through 2016: Changes in Soil Amended with Phosphate Fertilizer and Compost. Sci. Total Environ. 2021, 762, 143132. [Google Scholar] [CrossRef] [PubMed]
- Shaari, N.E.M.; Tajudin, M.T.F.M.; Khandaker, M.M.; Majrashi, A.; Alenazi, M.M.; Abdullahi, U.A.; Mohd, K.S. Cadmium Toxicity Symptoms and Uptake Mechanism in Plants: A Review. Braz. J. Biol. 2024, 84, e252143. [Google Scholar] [CrossRef]
- Chen, Y.; Chao, Z.F.; Jin, M.; Wang, Y.L.; Li, Y.; Wu, J.C.; Xiao, Y.; Peng, Y.; Lv, Q.Y.; Gui, S.; et al. A Heavy Metal Transporter Gene ZmHMA3a Promises Safe Agricultural Production on Cadmium-Polluted Arable Land. J. Genet. Genom. 2023, 50, 130–134. [Google Scholar] [CrossRef]
- Haider, F.U.; Liqun, C.; Coulter, J.A.; Cheema, S.A.; Wu, J.; Zhang, R.; Wenjun, M.; Farooq, M. Cadmium Toxicity in Plants: Impacts and Remediation Strategies. Ecotoxicol. Environ. Saf. 2021, 211, 111887. [Google Scholar] [CrossRef]
- Matayoshi, C.L.; Pena, L.B.; Arbona, V.; Gómez-Cadenas, A.; Gallego, S.M. Biochemical and Hormonal Changes Associated with Root Growth Restriction under Cadmium Stress during Maize (Zea mays L.) Pre-Emergence. Plant Growth Regul. 2022, 96, 269–281. [Google Scholar] [CrossRef]
- Baryla, A.; Carrier, P.; Franck, F.; Coulomb, C.; Sahut, C.; Havaux, M. Leaf Chlorosis in Oilseed Rape Plants (Brassica napus) Grown on Cadmium-Polluted Soil: Causes and Consequences for Photosynthesis and Growth. Planta 2001, 212, 696–709. [Google Scholar] [CrossRef]
- Waheed, A.; Haxim, Y.; Islam, W.; Ahmad, M.; Ali, S.; Wen, X.; Khan, K.A.; Ghramh, H.A.; Zhang, Z.; Zhang, D. Impact of Cadmium Stress on Growth and Physio-Biochemical Attributes of Eruca Sativa Mill. Plants 2022, 11, 2981. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Rizwan, M.; Ali, S.; Adrees, M.; Zia-ur-Rehman, M.; Qayyum, M.F.; Ok, Y.S.; Murtaza, G. Effect of Biochar on Alleviation of Cadmium Toxicity in Wheat (Triticum aestivum L.) Grown on Cd-Contaminated Saline Soil. Environ. Sci. Pollut. Res. 2018, 25, 25668–25680. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, F. bo Breeding for Low Cadmium Accumulation Cereals. J. Zhejiang Univ. Sci. B 2020, 21, 442–459. [Google Scholar] [CrossRef]
- Huang, X.; Duan, S.; Wu, Q.; Yu, M.; Shabala, S. Reducing Cadmium Accumulation in Plants: Structure–Function Relations and Tissue-Specific Operation of Transporters in the Spotlight. Plants 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.F.; Lestari, P.; Lee, K.J.; Kim, M.Y.; Lee, S.H.; Lee, B.W. Identification of Quantitative Trait Loci for Cadmium Accumulation and Distribution in Rice (Oryza sativa). Genome 2013, 56, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ritchie, M.D. From GWAS to Gene: Transcriptome-Wide Association Studies and Other Methods to Functionally Understand GWAS Discoveries. Front. Genet. 2021, 12, 713230. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, L.; Cao, Y.; Liu, Y.; Li, Y.; Wu, W.; Lan, Y.; Jiang, Y.; Gao, S.; Zhang, Z.; et al. Genome-Wide Association Analysis and QTL Mapping Reveal the Genetic Control of Cadmium Accumulation in Maize Leaf. BMC Genom. 2018, 19, 91. [Google Scholar] [CrossRef]
- Baseggio, M.; Murray, M.; Wu, D.; Ziegler, G.; Kaczmar, N.; Chamness, J.; Hamilton, J.P.; Robin Buell, C.; Vatamaniuk, O.K.; Buckler, E.S.; et al. Genome-Wide Association Study Suggests an Independent Genetic Basis of Zinc and Cadmium Concentrations in Fresh Sweet Corn Kernels. G3 Genes Genomes Genet. 2021, 11, jkab186. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, N.; Bao, F.; Zhao, G.; Jing, L.; Wang, G.; Han, Q.; Hao, Z.; Chen, B. Genome-Wide Association Study Reveals Genetic Basis of Trace Elements Accumulation in Maize Kernels. Agriculture 2022, 12, 262. [Google Scholar] [CrossRef]
- Yan, H.; Guo, H.; Xu, W.; Dai, C.; Kimani, W.; Xie, J.; Zhang, H.; Li, T.; Wang, F.; Yu, Y.; et al. GWAS-Assisted Genomic Prediction of Cadmium Accumulation in Maize Kernel with Machine Learning and Linear Statistical Methods. J. Hazard. Mater. 2023, 441, 129929. [Google Scholar] [CrossRef]
- Paape, T.; Heiniger, B.; Santo Domingo, M.; Clear, M.R.; Lucas, M.M.; Pueyo, J.J. Genome-Wide Association Study Reveals Complex Genetic Architecture of Cadmium and Mercury Accumulation and Tolerance Traits in Medicago Truncatula. Front. Plant Sci. 2022, 12, 806949. [Google Scholar] [CrossRef]
- Zhu, H.; Lai, R.; Chen, W.; Lu, C.; Chachar, Z.; Lu, S.; Lin, H.; Fan, L.; Hu, Y.; An, Y.; et al. Genetic Dissection of Maize (Zea mays L.) Trace Element Traits Using Genome-Wide Association Studies. BMC Plant Biol. 2023, 23, 631. [Google Scholar] [CrossRef]
- Liu, H.; Shi, J.; Sun, C.; Gong, H.; Fan, X.; Qiu, F.; Huang, X.; Feng, Q.; Zheng, X.; Yuan, N.; et al. Gene Duplication Confers Enhanced Expression of 27-KDa γ-Zein for Endosperm Modification in Quality Protein Maize. Proc. Natl. Acad. Sci. USA 2016, 113, 4964–4969. [Google Scholar] [CrossRef]
- Sahu, H.; Amadabade, J.; Dhiri, N. Association Mapping: A Useful Tool. Int. J. Plant Sci. 2015, 10, 85–94. [Google Scholar] [CrossRef]
- Ray, A.M.; Zuhlke, K.A.; Levin, A.M.; Douglas, J.A.; Cooney, K.A.; Petros, J.A. Sequence Variation in the Mitochondrial Gene Cytochrome c Oxidase Subunit I and Prostate Cancer in African American Men. Prostate 2009, 69, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Jiang, J.; Wang, J.; Du, J.; Lai, J.; Yang, C. Functional Characterization of Three Maize SIZ/PIAS-Type SUMO E3 Ligases. J. Plant Physiol. 2022, 268, 153588. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Qian, N.; Zhu, X.; Chen, H.; Wang, S.; Mei, H.; Zhang, Y. Variations and Transmission of QTL Alleles for Yield and Fiber Qualities in Upland Cotton Cultivars Developed in China. PLoS ONE 2013, 8, e57220. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Lai, R.; Xia, Y.; Li, R.; Zhao, W.; Siddique, K.H.M.; Chen, J.; Guo, P. Genetic Dissection of Tobacco (Nicotiana tabacum L.) Plant Height Using Single-Locus and Multi-Locus Genome-Wide Association Studies. Agronomy 2022, 12, 1047. [Google Scholar] [CrossRef]
- Feng, K.; Li, J.; Yang, Y.; Li, Z.; Wu, W. Cadmium Absorption in Various Genotypes of Rice under Cadmium Stress. Int. J. Mol. Sci. 2023, 24, 8019. [Google Scholar] [CrossRef]
- Li, X.; Du, J.; Sun, L.; Zhang, Y.; Feng, Y.; Zheng, L.; Wang, G.; Huang, X. Derivation of Soil Criteria of Cadmium for Safe Rice Production Applying Soil–Plant Transfer Model and Species Sensitivity Distribution. Int. J. Env. Res. Public Health 2022, 19, 8854. [Google Scholar] [CrossRef]
- Zhou, G.; Zhu, Q.; Mao, Y.; Chen, G.; Xue, L.; Lu, H.; Shi, M.; Zhang, Z.; Song, X.; Zhang, H.; et al. Multi-Locus Genome-Wide Association Study and Genomic Selection of Kernel Moisture Content at the Harvest Stage in Maize. Front. Plant Sci. 2021, 12, 697688. [Google Scholar] [CrossRef]
- Grapes, L.; Dekkers, J.C.M.; Rothschild, M.F.; Fernando, R.L. Comparing Linkage Disequilibrium-Based Methods for Fine Mapping Quantitative Trait Loci. Genetics 2004, 166, 1561–1570. [Google Scholar] [CrossRef]
- Wang, C.; Han, B. Twenty Years of Rice Genomics Research: From Sequencing and Functional Genomics to Quantitative Genomics. Mol. Plant 2022, 15, 593–619. [Google Scholar] [CrossRef]
- Li, Y.; Ding, L.; Zhou, M.; Chen, Z.; Ding, Y.; Zhu, C. Transcriptional Regulatory Network of Plant Cadmium Stress Response. Int. J. Mol. Sci. 2023, 24, 4378. [Google Scholar] [CrossRef] [PubMed]
- Sferra, G.; Fantozzi, D.; Scippa, G.S.; Trupiano, D. Key Pathways and Genes of Arabidopsis Thaliana and Arabidopsis Halleri Roots under Cadmium Stress Responses: Differences and Similarities. Plants 2023, 12, 1793. [Google Scholar] [CrossRef] [PubMed]
- Feki, K.; Tounsi, S.; Mrabet, M.; Mhadhbi, H.; Brini, F. Recent Advances in Physiological and Molecular Mechanisms of Heavy Metal Accumulation in Plants. Environ. Sci. Pollut. Res. 2021, 28, 64967–64986. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.L.; Zheng, M.M.; Tan, A.J.; Liu, Y.T.; Feng, D.; Lv, S.M. Research on the Mechanisms of Plant Enrichment and Detoxification of Cadmium. Biology 2021, 10, 544. [Google Scholar] [CrossRef] [PubMed]
- Moravčíková, D.; Žiarovská, J. The Effect of Cadmium on Plants in Terms of the Response of Gene Expression Level and Activity. Plants 2023, 12, 1848. [Google Scholar] [CrossRef]
- Li, C.; Hong, Y.; Sun, J.; Wang, G.; Zhou, H.; Xu, L.; Wang, L.; Xu, G. Temporal Transcriptome Analysis Reveals Several Key Pathways Involve in Cadmium Stress Response in Nicotiana tabacum L. Front. Plant Sci. 2023, 14, 1143349. [Google Scholar] [CrossRef]
- Niekerk, L.A.; Carelse, M.F.; Bakare, O.O.; Mavumengwana, V.; Keyster, M.; Gokul, A. The Relationship between Cadmium Toxicity and the Modulation of Epigenetic Traits in Plants. Int. J. Mol. Sci. 2021, 22, 7046. [Google Scholar] [CrossRef]
- Wang, Q.; Zeng, X.; Song, Q.; Sun, Y.; Feng, Y.; Lai, Y. Identification of Key Genes and Modules in Response to Cadmium Stress in Different Rice Varieties and Stem Nodes by Weighted Gene Co-Expression Network Analysis. Sci. Rep. 2020, 10, 9525. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Environment | Range | Mean | CV/% | Fg | Fe | Fg×e | H2/% |
|---|---|---|---|---|---|---|---|---|
| 2013 | HN | 0~0.06295 | 0.0068 | 186.68 | 23.27 * | 1086.34 * | 6.39 * | 75.92 |
| 2020 | JMO | 0~0.167 | 0.0286 | 88.23 | ||||
| 2021 | JMT | 0.0057~0.1711 | 0.0311 | 76.31 |
| Direction | P1 | P2 | Number of P1-Excellent Haplotype/ Allelic Variation | Number of P2-Excellent Haplotype/ Allelic Variation | Expected Offspring Haplotype/ Allelic Variation |
|---|---|---|---|---|---|
| Low Cd and relative high Mn/Fe | CAU95 * | CAU65 # | 11/2 | 10/2 | 12/2 |
| CAU95 * | CAU266 @ | 11/2 | 12/2 | 12/2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, R.; Xue, X.; Chachar, Z.; Zhu, H.; Chen, W.; Li, X.; Hu, Y.; Chen, M.; Zhang, X.; Li, J.; et al. Mapping Novel Loci and Candidate Genes Associated with Cadmium Content in Maize Using Genome-Wide Association Analysis. Agriculture 2025, 15, 389. https://doi.org/10.3390/agriculture15040389
Lai R, Xue X, Chachar Z, Zhu H, Chen W, Li X, Hu Y, Chen M, Zhang X, Li J, et al. Mapping Novel Loci and Candidate Genes Associated with Cadmium Content in Maize Using Genome-Wide Association Analysis. Agriculture. 2025; 15(4):389. https://doi.org/10.3390/agriculture15040389
Chicago/Turabian StyleLai, Ruiqiang, Xiaoming Xue, Zaid Chachar, Hang Zhu, Weiwei Chen, Xuhui Li, Yuanqiang Hu, Ming Chen, Xiangbo Zhang, Jiajia Li, and et al. 2025. "Mapping Novel Loci and Candidate Genes Associated with Cadmium Content in Maize Using Genome-Wide Association Analysis" Agriculture 15, no. 4: 389. https://doi.org/10.3390/agriculture15040389
APA StyleLai, R., Xue, X., Chachar, Z., Zhu, H., Chen, W., Li, X., Hu, Y., Chen, M., Zhang, X., Li, J., Fan, L., & Qi, Y. (2025). Mapping Novel Loci and Candidate Genes Associated with Cadmium Content in Maize Using Genome-Wide Association Analysis. Agriculture, 15(4), 389. https://doi.org/10.3390/agriculture15040389