
Construction of an SNP Fingerprinting Database and Population Genetic Analysis of Auricularia heimuer

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
Test Strain
2.2. Methods
2.2.1. DNA Extraction and Library Construction
2.2.2. Variant Detection and Annotation
2.2.3. Genetic Diversity and Population Structure Analysis
2.2.4. Design of KASP Markers and Genotyping
2.2.5. DNA Fingerprinting Map Construction
3. Results
3.1. Quality Control and Alignment Analysis of Resequencing Data
3.2. SNP Detection and Annotation
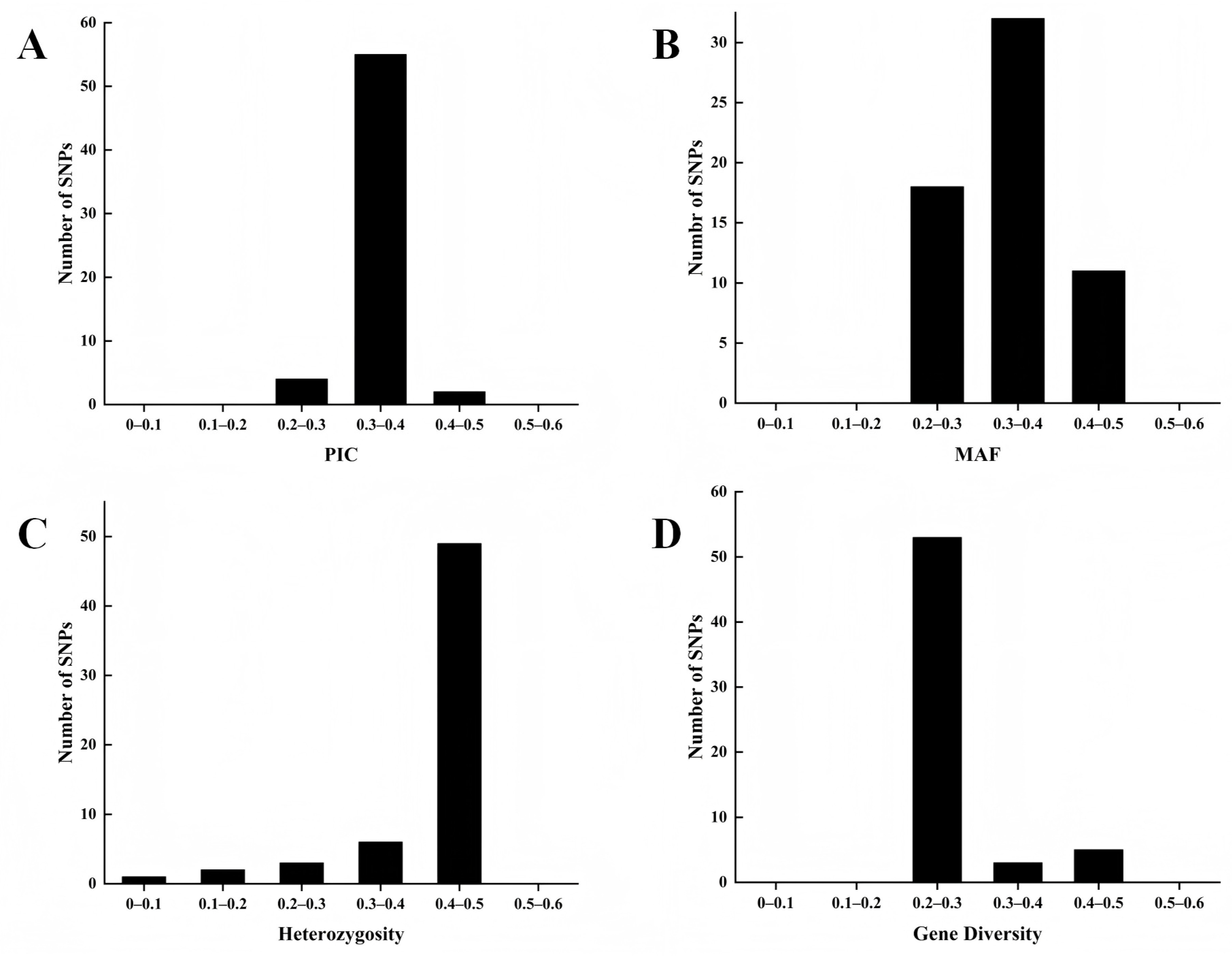
3.3. Genetic Diversity and Population Structure Analysis
3.4. Conversion and Selection of KASP Markers
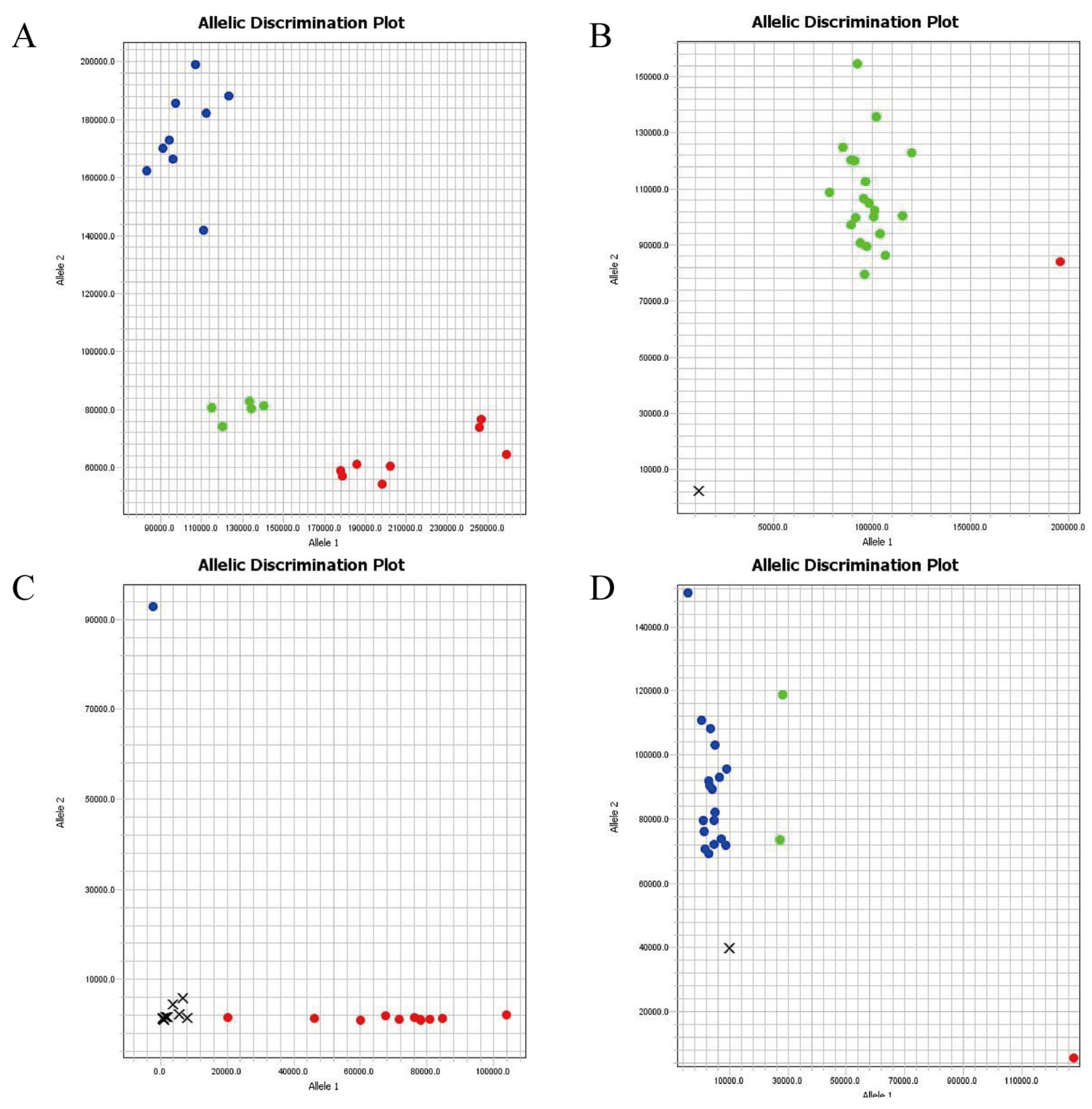
3.5. Genotyping Validation of KASP Markers
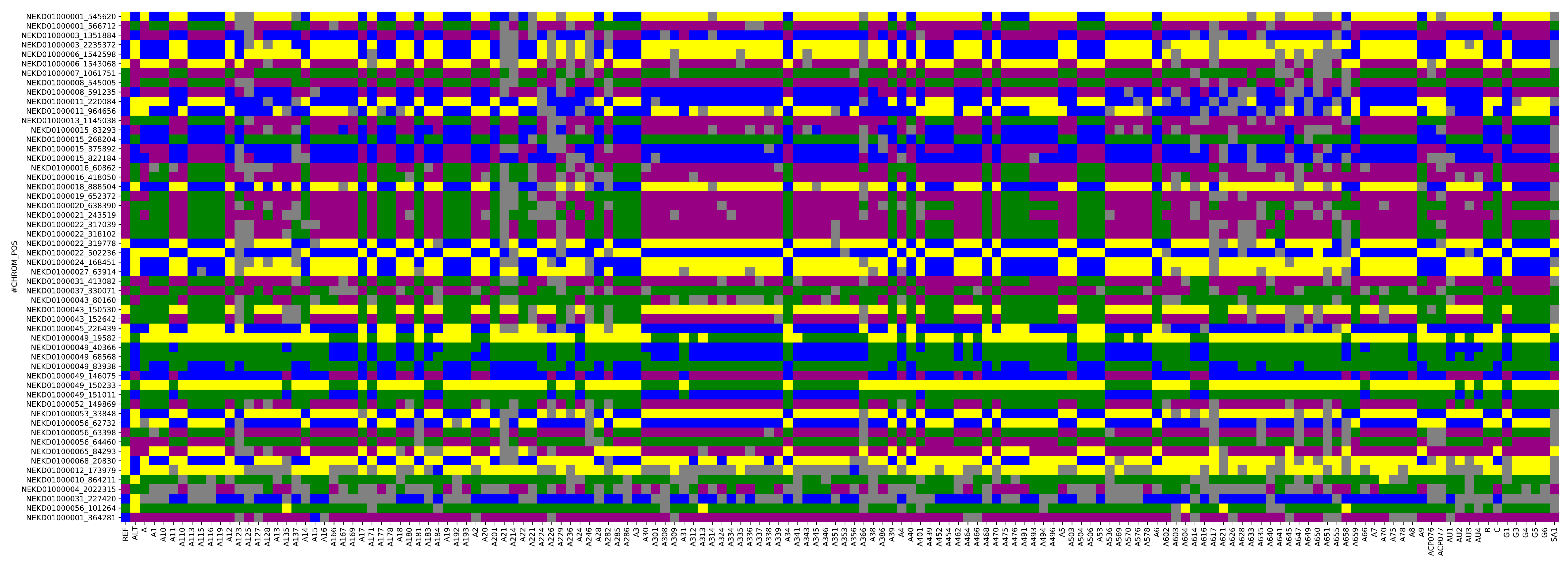
3.6. Construction of DNA Fingerprinting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Yuan, Y.; Malysheva, V.F.; Du, P.; Dai, Y.-C. Species clarification of the most important and cultivated Auricularia mushroom “Heimuer”: Evidence from morphological and molecular data. Phytotaxa 2014, 186, 241–253. [Google Scholar] [CrossRef]
- Islam, T.; Yao, F.; Kang, W.; Lu, L.; Xu, B. A systematic study on mycochemical profiles, antioxidant, and anti-inflammatory activities of 30 varieties of Jew’s ear (Auricularia auricula-judae). Food Sci. Hum. Wellness 2022, 11, 781–794. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, Y.; Jiang, K. Antioxidant activity in vitro and in vivo of the polysaccharides from different varieties of Auricularia auricula. Food Funct. 2016, 7, 3868–3879. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Yu, X.; Xu, B. Phenolic profiles, antioxidant capacities and metal chelating ability of edible mushrooms commonly consumed in China. LWT-Food Sci. Technol. 2016, 72, 423–431. [Google Scholar] [CrossRef]
- Yoon, S.-J.; Yu, M.-A.; Pyun, Y.-R.; Hwang, J.-K.; Chu, D.-C.; Juneja, L.R.; Mourão, P.A.S. The nontoxic mushroom Auricularia auricula contains a polysaccharide with anticoagulant activity mediated by antithrombin. Thromb. Res. 2003, 112, 151–158. [Google Scholar] [CrossRef]
- Yao, F.; Zhang, Y.; Lu, L.; Fang, M. Research progress on genetics and breeding of Auricularia auricula-judae. J. Fungal Res. 2015, 13, 125–128+122. [Google Scholar] [CrossRef]
- Oliveira, M.; Azevedo, L. Molecular Markers: An Overview of Data Published for Fungi over the Last Ten Years. J. Fungi 2022, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Das, S.P.; Choudhury, B.U.; Kumar, A.; Prakash, N.R.; Verma, R.; Chakraborti, M.; Devi, A.G.; Bhattacharjee, B.; Das, R.; et al. Advances in genomic tools for plant breeding: Harnessing DNA molecular markers, genomic selection, and genome editing. Biol. Res. 2024, 57, 80. [Google Scholar] [CrossRef]
- Tsykun, T.; Rellstab, C.; Dutech, C.; Sipos, G.; Prospero, S. Comparative assessment of SSR and SNP markers for inferring the population genetic structure of the common fungus Armillaria cepistipes. Heredity 2017, 119, 371–380. [Google Scholar] [CrossRef]
- Li, H.; Shi, L.; Tang, W.; Xia, W.; Zhong, Y.; Xu, X.; Xie, B.; Tao, Y. Comprehensive Genetic Analysis of Monokaryon and Dikaryon Populations Provides Insight Into Cross-Breeding of Flammulina filiformis. Front. Microbiol. 2022, 13, 887259. [Google Scholar] [CrossRef]
- Jördens, R. Progress of plant variety protection based on the International Convention for the Protection of New Varieties of Plants (UPOV Convention). World Pat. Inf. 2005, 27, 232–243. [Google Scholar] [CrossRef]
- Dipta, B.; Sood, S.; Mangal, V.; Bhardwaj, V.; Thakur, A.K.; Kumar, V.; Singh, B. KASP: A high-throughput genotyping system and its applications in major crop plants for biotic and abiotic stress tolerance. Mol. Biol. Rep. 2024, 51, 508. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Babu, R.; Hearne, S.; Olsen, M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): Overview of the technology and its application in crop improvement. Mol. Breed. 2014, 33, 1–14. [Google Scholar] [CrossRef]
- Wang, F.-q.; Fan, X.-c.; Zhang, Y.; Sun, L.; Liu, C.-h.; Jiang, J.-f. Establishment and application of an SNP molecular identification system for grape cultivars. J. Integr. Agric. 2022, 21, 1044–1057. [Google Scholar] [CrossRef]
- Yang, Y.; Lyu, M.; Liu, J.; Wu, J.; Wang, Q.; Xie, T.; Li, H.; Chen, R.; Sun, D.; Yang, Y.; et al. Construction of an SNP fingerprinting database and population genetic analysis of 329 cauliflower cultivars. BMC Plant Biol. 2022, 22, 522. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, H.; Xiang, X.; Yang, A.; Feng, Q.; Dai, P.; Li, Y.; Jiang, X.; Liu, G.; Zhang, X. Construction of a SNP Fingerprinting Database and Population Genetic Analysis of Cigar Tobacco Germplasm Resources in China. Front. Plant Sci. 2021, 12, 618133. [Google Scholar] [CrossRef]
- Grewal, S.; Hubbart-Edwards, S.; Yang, C.; Devi, U.; Baker, L.; Heath, J.; Ashling, S.; Scholefield, D.; Howells, C.; Yarde, J.; et al. Rapid identification of homozygosity and site of wild relative introgressions in wheat through chromosome-specific KASP genotyping assays. Plant Biotechnol. J. 2020, 18, 743–755. [Google Scholar] [CrossRef]
- Tang, W.; Lin, J.; Wang, Y.; An, H.; Chen, H.; Pan, G.; Zhang, S.; Guo, B.; Yu, K.; Li, H.; et al. Selection and Validation of 48 KASP Markers for Variety Identification and Breeding Guidance in Conventional and Hybrid Rice (Oryza sativa L.). Rice 2022, 15, 48. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Zhang, L.; Luo, J.; Zhao, H.; Zhang, J.; Wen, C. A new SNP genotyping technology Target SNP-seq and its application in genetic analysis of cucumber varieties. Sci. Rep. 2020, 10, 5623, Erratum in Sci. Rep. 2021, 11, 8010. https://doi.org/10.1038/s41598-021-86981-x. [Google Scholar] [CrossRef]
- Diao, B.; Xu, Z.; Liu, M.; Zhang, G.; Wang, G.; Zhang, Y.; Tian, X. Establishment and application of a SNP molecular identification system in Grifola frondosa. Front. Microbiol. 2024, 15, 1417014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Q.; Zhao, J.; Tang, K.; Lin, J.; Yin, Y. Comparison of rapid DNA extraction methods applied to PCR identification of medicinal mushroom Ganoderma spp. Prep. Biochem. Biotechnol. 2007, 37, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Xu, C.; Ren, Y.; Jian, Y.; Guo, Z.; Zhang, Y.; Xie, C.; Fu, J.; Wang, H.; Wang, G.; Xu, Y.; et al. Development of a maize 55 K SNP array with improved genome coverage for molecular breeding. Mol. Breed. 2017, 37, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Zhu, C.; Barkley, N.A.; Chen, Z.; Erpelding, J.E.; Murray, S.C.; Tuinstra, M.R.; Tesso, T.; Pederson, G.A.; Yu, J. Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection. Theor. Appl. Genet. 2009, 120, 13–23. [Google Scholar] [CrossRef]
- Cibulskis, K.; McKenna, A.; Fennell, T.; Banks, E.; DePristo, M.; Getz, G. ContEst: Estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 2011, 27, 2601–2602. [Google Scholar] [CrossRef]
- Jiao, L.; Han, C.; Zhu, J.; Zhang, P.; Ma, Y.; Dai, X.; Zhang, Y. Transcriptome analysis and development of EST-SSR markers in the mushroom Auricularia heimuer. Sci. Rep. 2024, 14, 12340. [Google Scholar] [CrossRef]
- Sun, X.; Yang, C.; Ma, Y.; Zhang, J.; Wang, L. Research progress of Auricularia heimuer on cultivation physiology and molecular biology. Front. Microbiol. 2022, 13, 1048249. [Google Scholar] [CrossRef]
- Du, P.; Cui, B.-K.; Zhang, C.-F.; Dai, Y.-C. Genetic diversity of wild Auricularia auricula-judae revealed by ISSR analysis. Biochem. Syst. Ecol. 2013, 48, 199–205. [Google Scholar] [CrossRef]
- Yao, F.-J.; Lu, L.-X.; Wang, P.; Fang, M.; Zhang, Y.-M.; Chen, Y.; Zhang, W.-T.; Kong, X.-H.; Lu, J.; Honda, Y. Development of a Molecular Marker for Fruiting Body Pattern in Auricularia auricula-judae. Mycobiology 2018, 46, 72–78. [Google Scholar] [CrossRef]
- Gazendam, I.; Mojapelo, P.; Bairu, M.W. Potato Cultivar Identification in South Africa Using a Custom SNP Panel. Plants 2022, 11, 1546. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Gong, C.; Mai, P.; Li, Z.; Sun, B.; Li, T. Genetic diversity and population structure analysis of 418 tomato cultivars based on single nucleotide polymorphism markers. Front. Plant Sci. 2024, 15, 1445734. [Google Scholar] [CrossRef]
- Ongom, P.O.; Fatokun, C.; Togola, A.; Salvo, S.; Oyebode, O.G.; Ahmad, M.S.; Jockson, I.D.; Bala, G.; Boukar, O. Molecular Fingerprinting and Hybridity Authentication in Cowpea Using Single Nucleotide Polymorphism Based Kompetitive Allele-Specific PCR Assay. Front. Plant Sci. 2021, 12, 734117. [Google Scholar] [CrossRef]
- Yin, L.; Yao, F.; Shi, C.; Lu, L.; Wang, T.; Liu, W. Genetic diversity of wild Auricularia heimuer germplasm resources based on SSR markers. Acta Edulis Fungi 2022, 29, 1–9. [Google Scholar] [CrossRef]
- Meng, Q.; Xie, Z.; Jiang, Z.; Xu, H.; Guo, J.; Li, Y.; Wang, C.; Hu, Z.; Yan, S.; Liu, H. Population genetic structure analysis of the wild esculenta clade (yellow morels) of Morchella spp. in Qinghai. Mycosystema 2021, 40, 1328–1342+1370–1372. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Mun, J.-H.; Jeong, Y.-M.; Joo, S.-H.; Yu, H.-J. Assembly of a radish core collection for evaluation and preservation of genetic diversity. Hortic. Environ. Biotechnol. 2018, 59, 711–721. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker ID | Position | Ref | Alt | PIC | MAF | Heterozygosity | Gene Diversity |
|---|---|---|---|---|---|---|---|
| rs1 | 545,620 | C | T | 0.320 | 0.276 | 0.399 | 0.200 |
| rs2 | 566,712 | G | A | 0.321 | 0.279 | 0.402 | 0.201 |
| rs3 | 1,351,884 | G | T | 0.363 | 0.392 | 0.477 | 0.238 |
| rs4 | 2,235,372 | T | C | 0.363 | 0.390 | 0.476 | 0.338 |
| rs5 | 1,542,598 | T | C | 0.364 | 0.395 | 0.478 | 0.239 |
| rs6 | 1,543,068 | C | G | 0.363 | 0.392 | 0.477 | 0.238 |
| rs7 | 1,061,751 | A | G | 0.362 | 0.388 | 0.475 | 0.237 |
| rs8 | 545,005 | A | G | 0.365 | 0.403 | 0.481 | 0.241 |
| rs9 | 591,235 | G | T | 0.372 | 0.450 | 0.495 | 0.447 |
| rs10 | 220,084 | T | C | 0.360 | 0.381 | 0.472 | 0.236 |
| rs11 | 964,656 | T | C | 0.375 | 0.479 | 0.499 | 0.450 |
| rs12 | 1,145,038 | G | A | 0.354 | 0.359 | 0.460 | 0.230 |
| rs13 | 83,293 | T | G | 0.368 | 0.420 | 0.487 | 0.244 |
| rs14 | 268,204 | T | A | 0.355 | 0.362 | 0.462 | 0.231 |
| rs15 | 375,892 | G | T | 0.365 | 0.403 | 0.481 | 0.241 |
| rs16 | 822,184 | G | T | 0.331 | 0.299 | 0.419 | 0.309 |
| rs17 | 60,862 | G | A | 0.342 | 0.323 | 0.437 | 0.219 |
| rs18 | 418,050 | G | A | 0.324 | 0.284 | 0.407 | 0.203 |
| rs19 | 888,504 | T | C | 0.367 | 0.414 | 0.485 | 0.243 |
| rs20 | 652,372 | A | G | 0.321 | 0.279 | 0.402 | 0.301 |
| rs21 | 638,390 | G | A | 0.373 | 0.455 | 0.496 | 0.448 |
| rs22 | 243,519 | G | A | 0.338 | 0.315 | 0.432 | 0.216 |
| rs23 | 317,039 | G | A | 0.361 | 0.385 | 0.473 | 0.237 |
| rs24 | 318,102 | G | A | 0.364 | 0.396 | 0.478 | 0.239 |
| rs25 | 319,778 | C | T | 0.363 | 0.392 | 0.477 | 0.238 |
| rs26 | 502,236 | T | C | 0.357 | 0.370 | 0.466 | 0.233 |
| rs27 | 168,451 | T | C | 0.360 | 0.380 | 0.471 | 0.236 |
| rs28 | 63,914 | T | C | 0.353 | 0.354 | 0.457 | 0.229 |
| rs29 | 413,082 | A | G | 0.324 | 0.283 | 0.406 | 0.203 |
| rs30 | 330,071 | G | A | 0.373 | 0.458 | 0.497 | 0.248 |
| rs31 | 80,160 | A | G | 0.329 | 0.294 | 0.415 | 0.207 |
| rs32 | 150,530 | C | A | 0.361 | 0.385 | 0.473 | 0.237 |
| rs33 | 152,642 | G | A | 0.363 | 0.392 | 0.477 | 0.238 |
| rs34 | 226,439 | C | T | 0.329 | 0.293 | 0.415 | 0.207 |
| rs35 | 19,582 | C | A | 0.332 | 0.300 | 0.420 | 0.210 |
| rs36 | 40,366 | A | T | 0.358 | 0.373 | 0.468 | 0.234 |
| rs37 | 68,568 | A | T | 0.343 | 0.327 | 0.440 | 0.220 |
| rs38 | 83,938 | A | T | 0.329 | 0.293 | 0.415 | 0.207 |
| rs39 | 146,075 | T | G | 0.362 | 0.387 | 0.474 | 0.437 |
| rs40 | 150,233 | C | A | 0.322 | 0.280 | 0.403 | 0.202 |
| rs41 | 151,011 | A | T | 0.325 | 0.287 | 0.409 | 0.204 |
| rs42 | 149,869 | A | G | 0.370 | 0.430 | 0.490 | 0.245 |
| rs43 | 33,848 | T | C | 0.359 | 0.375 | 0.469 | 0.234 |
| rs44 | 62,732 | T | C | 0.351 | 0.347 | 0.453 | 0.227 |
| rs45 | 63,398 | G | A | 0.351 | 0.348 | 0.454 | 0.227 |
| rs46 | 64,460 | A | G | 0.348 | 0.339 | 0.448 | 0.224 |
| rs47 | 84,293 | C | G | 0.335 | 0.307 | 0.425 | 0.213 |
| rs48 | 20,830 | C | T | 0.339 | 0.317 | 0.433 | 0.217 |
| rs49 | 173,979 | C | T | 0.249 | 0.277 | 0.292 | 0.246 |
| rs50 | 864,211 | A | C | 0.128 | 0.422 | 0.137 | 0.269 |
| rs51 | 2,022,315 | G | A | 0.375 | 0.489 | 0.500 | 0.250 |
| rs52 | 227,420 | T | C | 0.251 | 0.393 | 0.295 | 0.247 |
| rs53 | 101,264 | A | C | 0.124 | 0.277 | 0.133 | 0.266 |
| rs54 | 364,281 | T | G | 0.093 | 0.293 | 0.098 | 0.249 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, K.; Feng, Q.; Yao, F.; Lu, L.; Fang, M.; Ma, X.; Sun, X. Construction of an SNP Fingerprinting Database and Population Genetic Analysis of Auricularia heimuer. Agriculture 2025, 15, 884. https://doi.org/10.3390/agriculture15080884
Shao K, Feng Q, Yao F, Lu L, Fang M, Ma X, Sun X. Construction of an SNP Fingerprinting Database and Population Genetic Analysis of Auricularia heimuer. Agriculture. 2025; 15(8):884. https://doi.org/10.3390/agriculture15080884
Chicago/Turabian StyleShao, Kaisheng, Qiuyu Feng, Fangjie Yao, Lixin Lu, Ming Fang, Xiaoxu Ma, and Xu Sun. 2025. "Construction of an SNP Fingerprinting Database and Population Genetic Analysis of Auricularia heimuer" Agriculture 15, no. 8: 884. https://doi.org/10.3390/agriculture15080884
APA StyleShao, K., Feng, Q., Yao, F., Lu, L., Fang, M., Ma, X., & Sun, X. (2025). Construction of an SNP Fingerprinting Database and Population Genetic Analysis of Auricularia heimuer. Agriculture, 15(8), 884. https://doi.org/10.3390/agriculture15080884