
A New Approach to Characterize Siderophore-Type Ligands in Seawater by Solid Phase Synthesis and SPE-HPLC-ESI-MS/MS Analysis
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Artificial Seawater
2.2. Siderophore-Type Standards
2.3. Central Composite Design
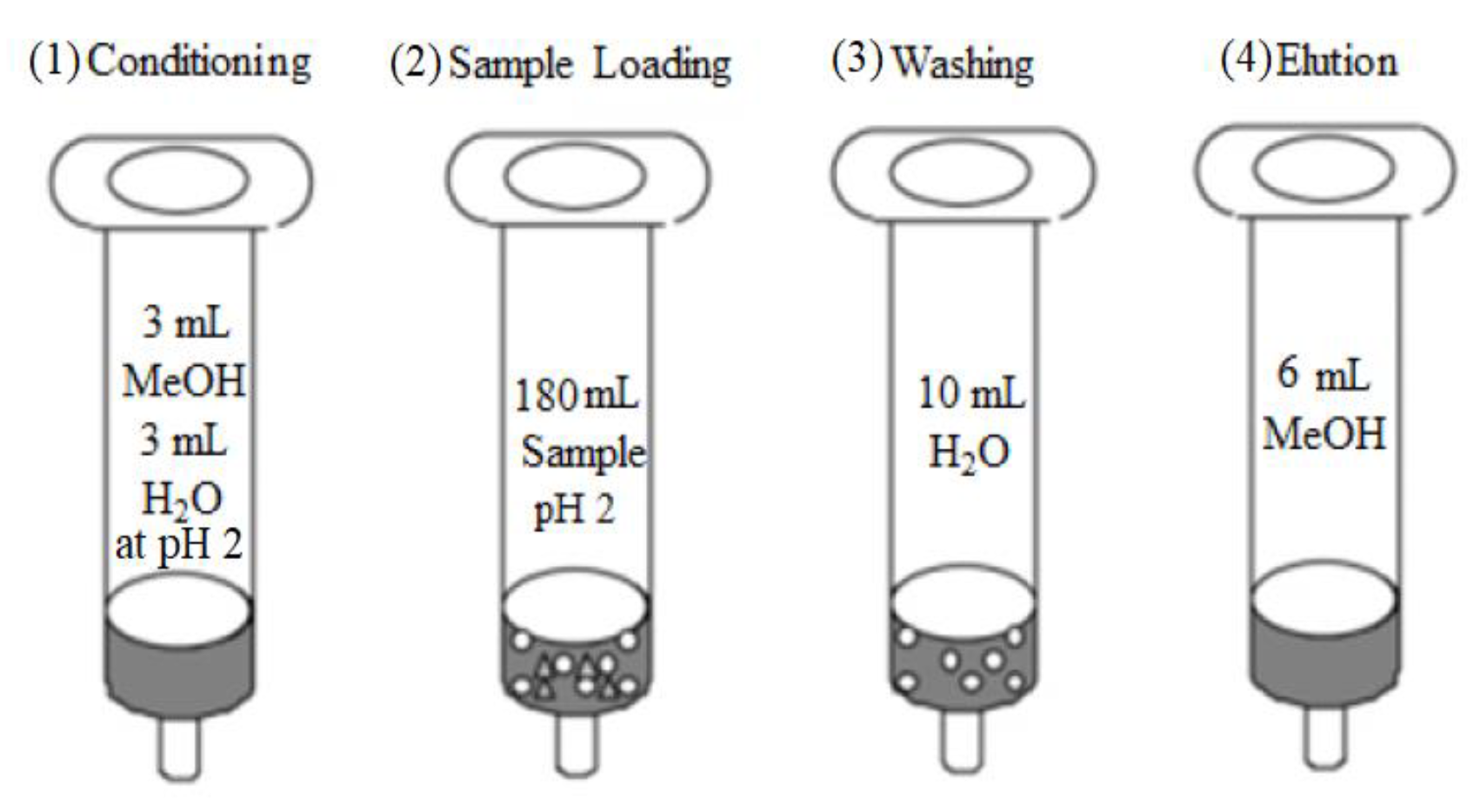
2.4. Extraction and Preconcentration Procedure
2.5. HPLC-ESI-MS/MS Conditions
3. Results
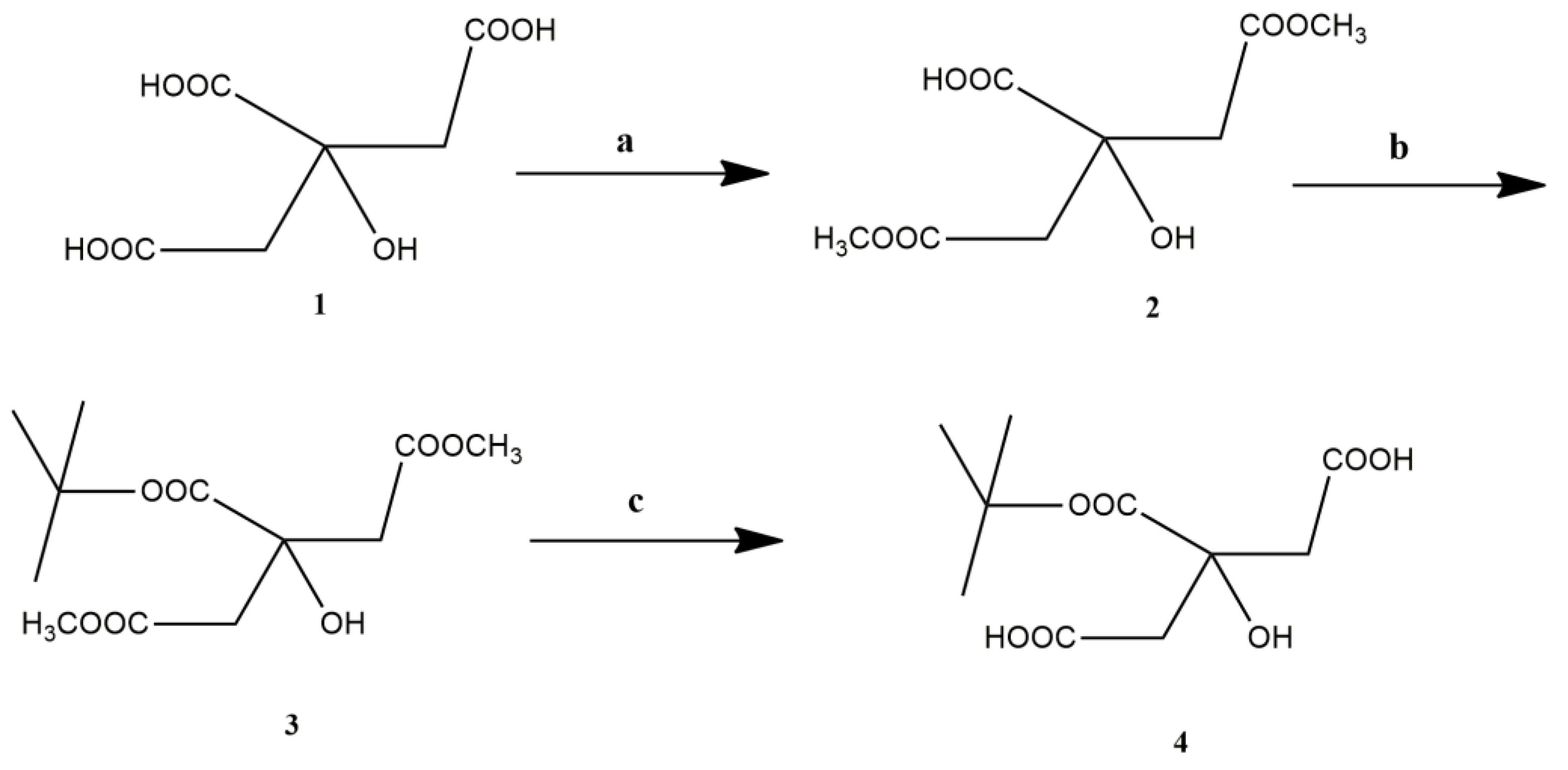
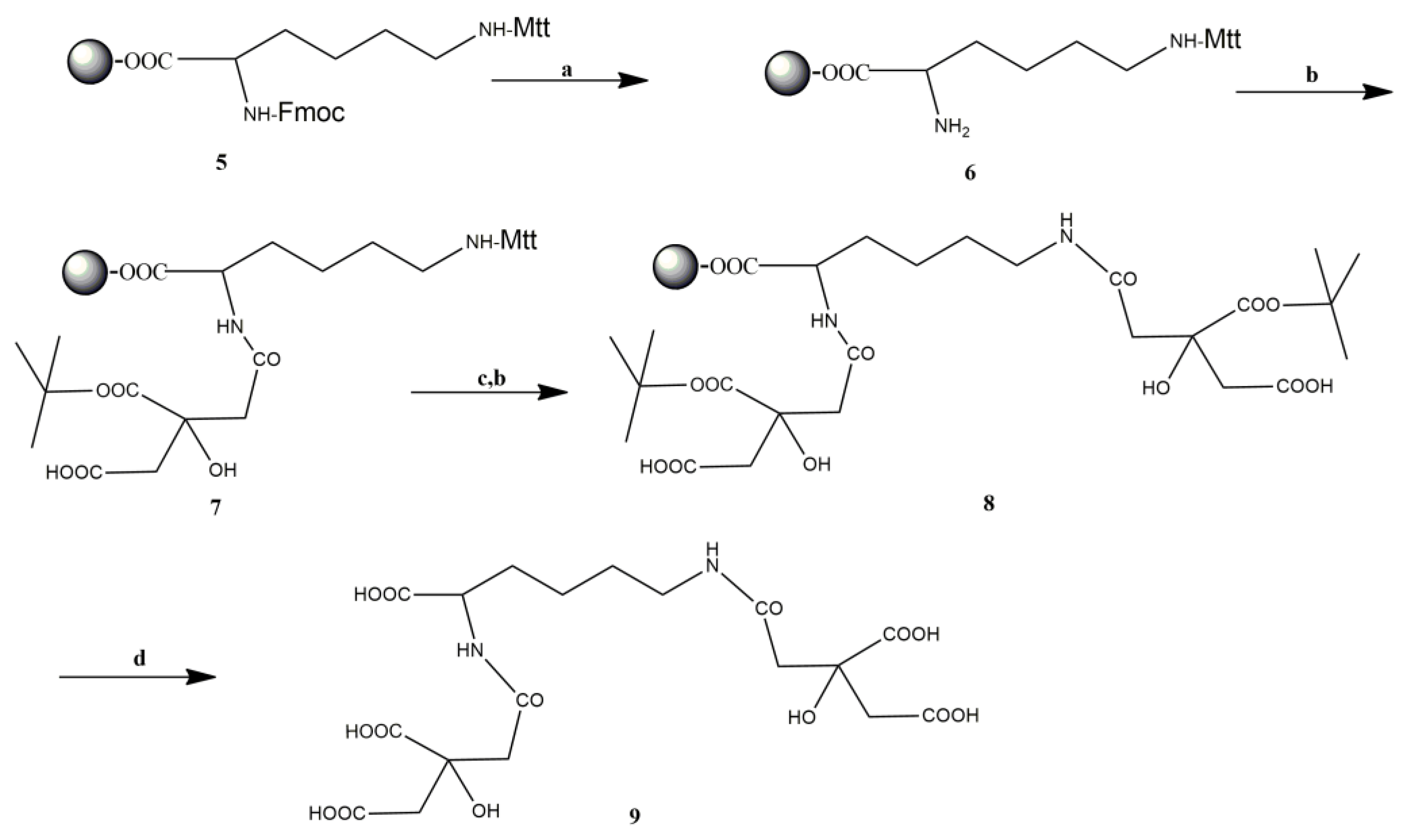
3.1. Synthesis
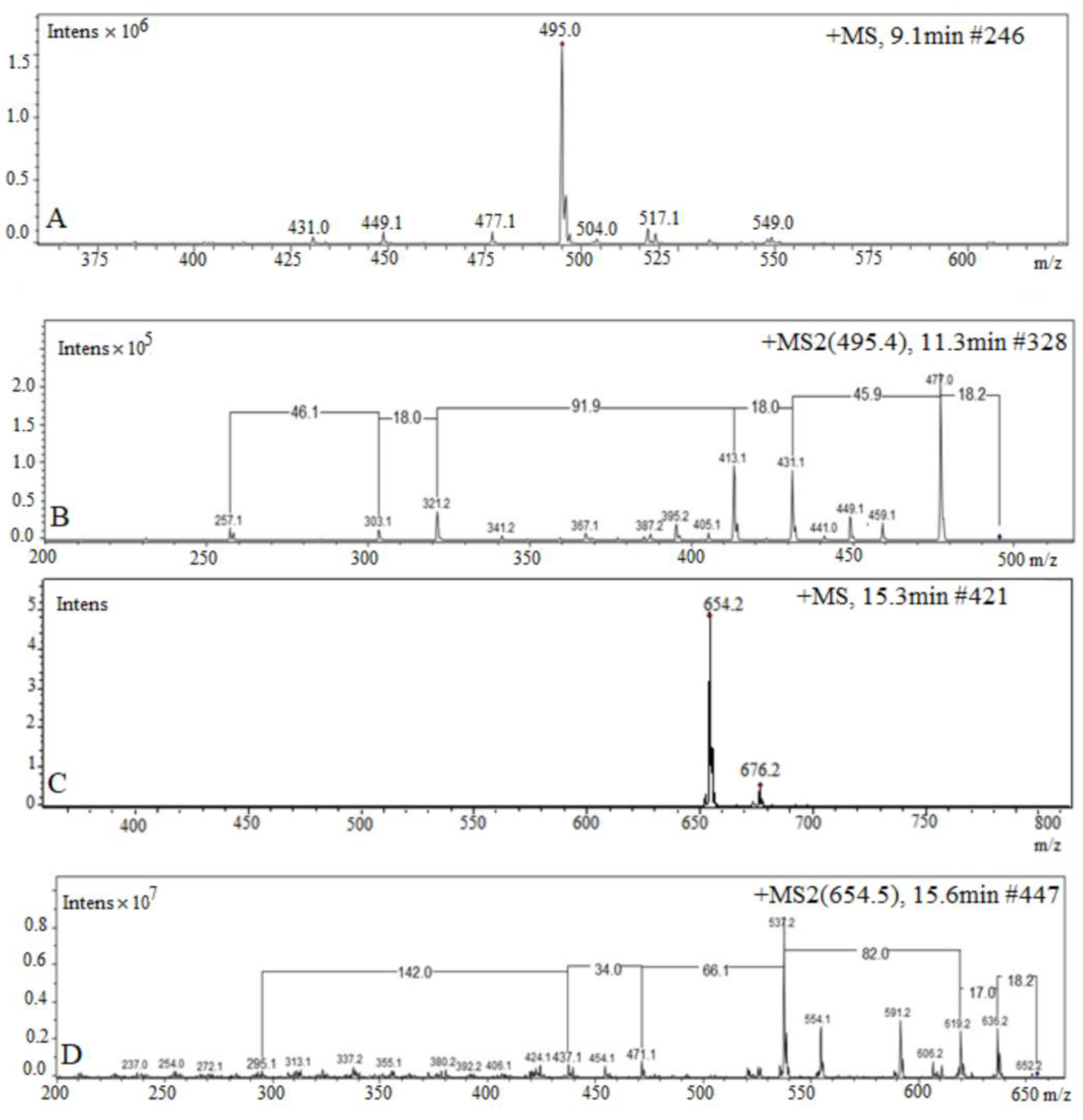
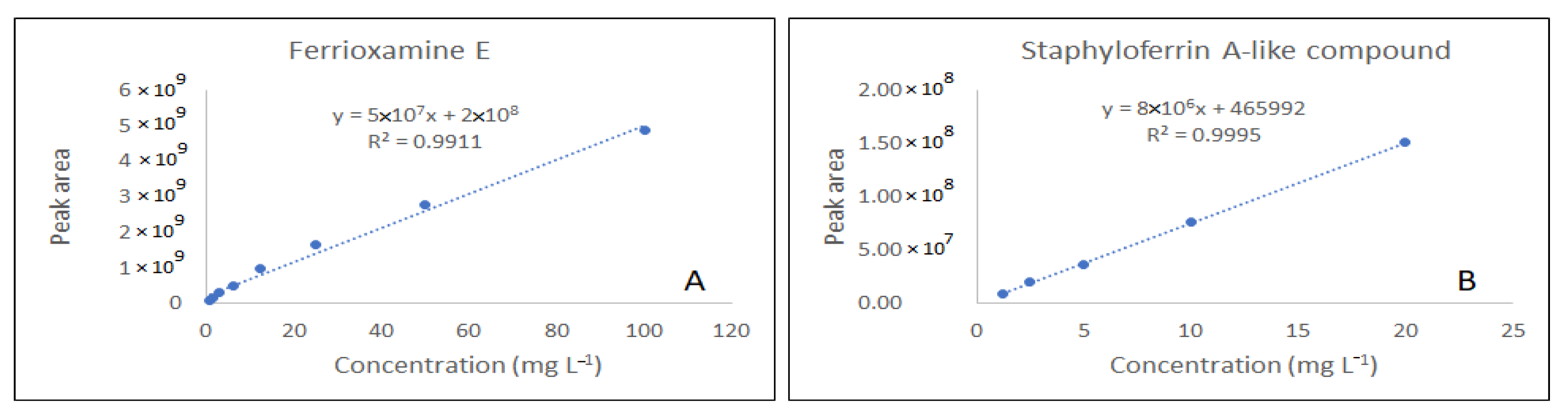
3.2. HPLC-ESI-MS/MS Analysis
3.3. Optimization of the Solid Phase Extraction by Central Composite Design
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Ibisanmi, E.; Sander, S.G.; Boyd, P.W.; Bowie, A.R.; Hunter, K.A. Vertical distributions of iron-(III) complexing ligands in the Southern Ocean. Deep Sea Res. II 2011, 58, 2113–2125. [Google Scholar] [CrossRef]
- Barbeau, K.; Rue, E.L.; Trick, C.G.; Bruland, K.W. Photochemical reactivity of siderophores produced by marine heterotrophic bacteria and cyanobacteria based on characteristic Fe(III) binding groups. Limnol. Oceanogr. 2003, 48, 1069–1078. [Google Scholar] [CrossRef]
- Gledhill, M.; Buck, K.N. The organic complexation of iron in the marine environment: A review. Front. Microbiol. 2012, 3, 69. [Google Scholar] [CrossRef]
- Rivaro, P.; Ardini, F.; Grotti, M.; Aulicino, G.; Cotroneo, Y.; Fusco, G.; Mangoni, O.; Bolinesi, F.; Saggiomo, M.; Celussi, M. Mesoscale variability related to iron speciation in a coastal Ross Sea area (Antarctica) during summer 2014. Chem. Ecol. 2019, 35, 1–19. [Google Scholar] [CrossRef]
- Reid, R.T.; Livet, D.H.; Faulkner, D.J.; Butler, A. A siderophore from a marine bacterium with an exceptional ferric ion affinity constant. Nature 1993, 366, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, Y.; Lu, Y.; Wang, B.; Sun, J.; Zhang, H.; Wang, H. Chemistry and biology of siderophores from marine microbes. Mar. Drugs 2019, 17, 562. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.A.; Boyd, P.W. Iron-binding ligands and their role in the ocean biogeochemistry of iron. Environ. Chem. 2007, 4, 221–232. [Google Scholar] [CrossRef]
- Vraspir, J.M.; Butler, A. Chemistry of marine ligands and siderophores. Annu. Rev. Mar. Sci. 2009, 64, 43–63. [Google Scholar] [CrossRef]
- Bundy, R.M.; Boiteau, R.M.; McLean, C.; Turk-Kubo, K.A.; McIlvin, M.R.; Saito, M.A.; Van Mooy, B.A.S.; Repeta, D.J. Distinct siderophores contribute to iron cycling in the mesopelagic at station ALOHA. Front. Mar. Sci. 2018, 5, 61. [Google Scholar] [CrossRef]
- Laglera, L.M.; Tovar-Sanchez, A.; Sukekava, C.F.; Naik, H.; Naqvi, S.W.A.; Wolf-Gladrow, D.A. Iron organic speciation during the LOHAFEX experiment: Iron ligands release under biomass control by copepod grazing. J. Mar. Syst. 2020, 207, 103151. [Google Scholar] [CrossRef]
- Ahmed, E.; Holmström, S.J.M. Siderophores in environmental research: Roles and applications. Microb. Biotechnol. 2014, 7, 196–208. [Google Scholar] [CrossRef]
- Boiteau, R.M.; Mende, D.R.; Hawco, N.J.; Mcilvin, M.R.; Fitzsimmons, J.N. Siderophore-based microbial adaptations to iron scarcity across the eastern Pacific Ocean. Proc. Natl. Acad. Sci. USA 2016, 13, 14237–14242. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.M.; Mills, M.M.; Arrigo, K.R.; Bopp, L.; Boyd, P.W.; Galbraith, E.D.; Geider, R.J.; Guieu, C.; Jaccard, S.L.; Jickells, T.D.; et al. Processes and patterns of oceanic nutrient limitation. Nature 2013, 6, 701–710. [Google Scholar] [CrossRef]
- De Serrano, L.O.; Camper, A.K.; Richards, A.M. An overview of siderophores for iron acquisition in microorganisms living in the extreme. Biometals 2016, 29, 551–571. [Google Scholar] [CrossRef]
- Boukhalfa, H.; Lack, J.; Reilly, S.D.; Hersman, L.; Neu, M.P. Siderophore production and facilitated uptake of iron and plutonium in P. putida. AIP Conf. Proc. 2003, 673, 343–344. [Google Scholar] [CrossRef]
- Zajdowicz, S.; Haller, J.C.; Krafft, A.E.; Hunsucker, S.W.; Mant, C.T.; Duncan, M.W.; Hodges, R.S.; Jones, D.N.M.; Holmes, R.K. Purification and structural characterization of siderophore (Corynebactin) from Corynebacterium diphtheriae. PLoS ONE 2012, 7, e34591. [Google Scholar] [CrossRef]
- McCormack, P.; Worsfold, P.J.; Gledhill, M. Separation and detection of siderophores produced by marine bacterioplankton using high-performance liquid chromatography with electrospray ionization mass spectrometry. Anal. Chem. 2003, 75, 2647–2652. [Google Scholar] [CrossRef]
- Mawji, E.; Gledhill, M.; Milton, J.A.; Tarran, G.A.; Ussher, S.; Thompson, A.; Wolff, G.A.; Worsfold, P.J.; Achterberg, E.P. Hydroxamate siderophores: Occurrence and importance in the Atlantic Ocean. Environ. Sci. Technol. 2008, 42, 8675–8680. [Google Scholar] [CrossRef]
- Dittmar, T.; Koch, B.; Hertkorn, N.; Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr. Methods 2008, 6, 230–235. [Google Scholar] [CrossRef]
- D’Andrilli, J.; Dittmar, T.; Koch, B.P.; Purcell, J.M.; Marshall, A.G.; Cooper, W.T. Comprehensive characterization of marine dissolved organic matter by Fourier transformation cyclotron resonance mass spectrometry with electrospray and atmospheric pressure photoionization. Rapid Commun. Mass Spectrom. 2010, 24, 643–650. [Google Scholar] [CrossRef]
- Velasquez, I.; Nunn, B.L.; Ibisanmi, E.; Goodlett, D.R.; Hunter, K.A.; Sander, S.G. Detection of hydroxamate siderophores in coastal and Sub-Antarctic waters off the Southeastern Coast of New Zealand. Mar. Chem. 2011, 126, 97–107. [Google Scholar] [CrossRef]
- Konetschny-Rapp, S.; Jung, G.; Meiwes, J.; Zahner, H. Staphyloferrin A: A structurally new siderophore from staphylococci. Eur. J. Biochem. 1990, 74, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Boiteau, R.M.; Fitzsimmons, J.N.; Repeta, D.J.; Boyle, E.A. Detection of iron ligands in seawater and marine cyanobacteria cultures by High-Performance Liquid Chromatography—Inductively Coupled Plasma-Mass Spectrometry. Anal. Chem. 2013, 85, 4357–4362. [Google Scholar] [CrossRef] [PubMed]
- Manck, L.E.; Park, J.; Tully, B.J.; Barbeau, K.A.; Poire, A.M.; Bundy, R.M.; Dupont, C.L. Petrobactin, a siderophore produced by Alteromonas, mediates community iron acquisition in the global ocean. ISME J. 2021, 16, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, I.B.; Ibisanmi, E.; Maas, E.W.; Boyd, P.W.; Nodder, S.; Sander, S.G. Ferrioxamine siderophores detected amongst iron binding ligands produced during the remineralization of marine particles. Front. Mar. Sci. 2016, 3, 172. [Google Scholar] [CrossRef]
- Budzikiewicz, H. Bacterial citrate siderophores. Mini-Rev. Org. Chem. 2005, 2, 119–124. [Google Scholar] [CrossRef]
- Berner, I.; Konetschny-Rapp, S.; Jung, G.; Winkelmann, G. Characterization of ferrioxamine E as the principal siderophore of Erwinia herbicola (Enterobacter agglomerans). Biol. Met. 1988, 1, 51–56. [Google Scholar] [CrossRef]
- Kingsley, R.A.; Reissbrodt, R.; Rabsch, W.; Ketley, J.M.; Tsolis, R.M.; Everest, P.; Dougan, G.; Bäumler, A.J.; Roberts, M.; Williams, P.H. Ferrioxamine-mediated Iron(III) utilization by Salmonella enterica. Appl. Environ. Microbiol. 1999, 65, 1610–1618. [Google Scholar] [CrossRef]
- Leardi, R.; Melzi, C.; Polotti, G. Chemometric Agile Tool (CAT). 2017. Available online: http://gruppochemiometria.it/index.php/software (accessed on 15 June 2023).
- Benedetti, B.; Caponigro, V.; Ardini, F. Experimental design step by step: A practical guide for beginners. Crit. Rev. Anal. Chem. 2022, 52, 1015–1028. [Google Scholar] [CrossRef]
- Pandey, R.K.; Jarvis, G.G.; Low, P.S. Chemical synthesis of staphyloferrin A and its application for Staphylococcus aureus detection. Org. Biomol. Chem. 2014, 12, 1707–1710. [Google Scholar] [CrossRef]
- Guo, H.; Naser, S.A.; Ghobrial, G.; Phanstiel, O. Synthesis and biological evaluation of new citrate-based siderophores as potential probes for the mechanism of iron uptake in mycobacteria. J. Med. Chem. 2002, 45, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Gledhill, M. Electrospray ionisation-mass spectrometry of hydroxamate siderophores. Analyst 2001, 126, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Sidebottom, A.M.; Karty, J.A.; Carlson, E.E. Accurate mass MS/MS/MS analysis of siderophores Ferrioxamine B and E1 by Collision-Induced Dissociation Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.L.; Price, N.M.; Bruland, K.W. Iron chemistry in seawater and its relationship to phytoplankton: A workshop report. Mar. Chem. 1995, 48, 157–182. [Google Scholar] [CrossRef]
- Macrellis, H.M.; Trick, C.G.; Rue, E.L.; Smith, G.; Bruland, K.W. Collection and detection of natural iron-binding ligands from seawater. Mar. Chem. 2001, 76, 175–187. [Google Scholar] [CrossRef]
- Achterberg, E.P.; Holland, T.W.; Bowie, A.R.; Mantoura, R.F.C.; Worsfold, P.J. Determination of iron in seawater. Anal. Chim. Acta 2001, 442, 1–14. [Google Scholar] [CrossRef]
- Vong, L.; Laës, A.; Blain, S. Determination of iron–porphyrin-like complexes at nanomolar levels in seawater. Anal. Chim. Acta 2007, 588, 237–244. [Google Scholar] [CrossRef]
- Hopkinson, B.M.; Morel, F.M.M. The role of siderophores in iron acquisition by photosynthetic marine microorganisms. BioMetals 2009, 22, 659–669. [Google Scholar] [CrossRef]
- Longhini, C.M.; Sá, F.; Neto, R.R. Review and synthesis: Iron input, biogeochemistry, and ecological approaches in seawater. Environ. Rev. 2018, 27, 125–137. [Google Scholar] [CrossRef]
- Butler, A. Marine siderophores and microbial iron mobilization. Biometals 2005, 18, 369–374. [Google Scholar] [CrossRef]
- Rivaro, P.; Ardini, F.; Grotti, M.; Vivado, D.; Salis, A.; Damonte, G. Detection of carbohydrates in sea ice extracellular polymeric substances via solid-phase extraction and HPLC-ESI-MS/MS. Mar. Chem. 2021, 228, 103911. [Google Scholar] [CrossRef]
- D’Andrilli, J.; Cooper, W.T.; Foreman, C.M.; Marshall, A.G. An ultrahigh-resolution mass spectrometry index to estimate natural organic matter lability. Rapid Commun. Mass Spectrom. 2015, 29, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, D.X.; Fang, K.; Liu, B.M. Determination of siderophores in seawater by high performance liquid chromatography-tandem mass spectrometry coupled with solid phase extraction. Chin. J. Anal. Chem. 2015, 43, 1285–1290. [Google Scholar] [CrossRef]
- Waska, H.; Koschinsky, A.; Ruiz Chancho, M.J.; Dittmar, T. Investigating the potential of solid-phase extraction and Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) for the isolation and identification of dissolved metal–organic complexes from natural waters. Mar. Chem. 2015, 173, 78–92. [Google Scholar] [CrossRef]
- Bhattacharya, S. Central Composite Design for Response Surface Methodology and Its Application in Pharmacy. In Response Surface Methodology in Engineering Science; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constituent | Molar Concentration (Mol L−1) | Weight (g) |
|---|---|---|
| NaCl | 0.47 | 109.87 |
| Na2SO4 × 10 H2O | 1.87 × 10−2 | 24.10 |
| KCl | 6.25 × 10−3 | 1.864 |
| CaCl2 × 2 H2O | 6.25 × 10−5 | 2.934 |
| NaHCO3 | 1.58 × 10−3 | 0.531 |
| KBr | 5.63 × 10−4 | 0.268 |
| H3BO3 | 3.83 × 10−4 | 0.080 |
| SrCl2 | 8.20 × 10−5 | 0.052 |
| Experiment | Factor 1 | Factor 2 | pH | Sample Volume (mL) |
|---|---|---|---|---|
| 1 | −1 | −1 | 2 | 50 |
| 2 | +1 | −1 | 5 | 50 |
| 3 | −1 | +1 | 2 | 200 |
| 4 | +1 | +1 | 5 | 200 |
| 5 | 0 | +1 | 3.5 | 200 |
| 6 | 0 | −1 | 3.5 | 50 |
| 7 | +1 | 0 | 5 | 125 |
| 8 | −1 | 0 | 2 | 125 |
| 9 | 0 | 0 | 3.5 | 125 |
| 10 | 0 | 0 | 3.5 | 125 |
| 11 | 0 | 0 | 3.5 | 125 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivaro, P.; Vivado, D.; Ianni, C.; Salis, A.; Parodi, A.; Millo, E. A New Approach to Characterize Siderophore-Type Ligands in Seawater by Solid Phase Synthesis and SPE-HPLC-ESI-MS/MS Analysis. J. Mar. Sci. Eng. 2024, 12, 110. https://doi.org/10.3390/jmse12010110
Rivaro P, Vivado D, Ianni C, Salis A, Parodi A, Millo E. A New Approach to Characterize Siderophore-Type Ligands in Seawater by Solid Phase Synthesis and SPE-HPLC-ESI-MS/MS Analysis. Journal of Marine Science and Engineering. 2024; 12(1):110. https://doi.org/10.3390/jmse12010110
Chicago/Turabian StyleRivaro, Paola, Davide Vivado, Carmela Ianni, Annalisa Salis, Alice Parodi, and Enrico Millo. 2024. "A New Approach to Characterize Siderophore-Type Ligands in Seawater by Solid Phase Synthesis and SPE-HPLC-ESI-MS/MS Analysis" Journal of Marine Science and Engineering 12, no. 1: 110. https://doi.org/10.3390/jmse12010110