
Development of Single-Nucleotide Polymorphism Markers and Population Genetic Analysis of the Hadal Amphipod Alicella gigantea across the Mariana and New Britain Trenches


Abstract
:1. Introduction
2. Materials and Methods
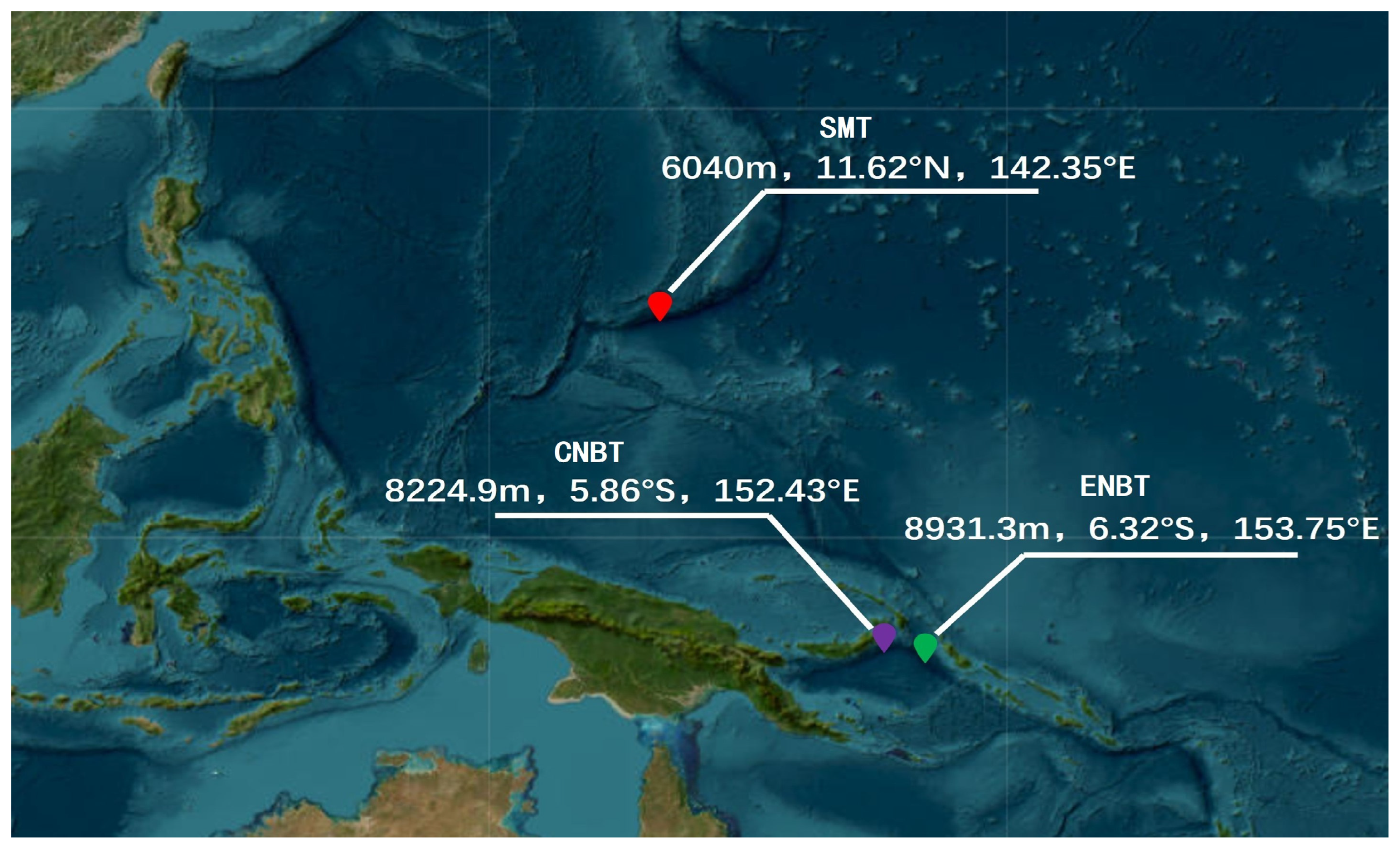
2.1. Sample Collection
2.2. SLAF Sequencing and Genotyping
2.3. Genetic Population Analysis Regarding the Three A. gigantea Geographic Populations
2.4. Environmental Adaptation Locus Screening and Functional Annotations
3. Results
3.1. SLAF Sequencing and Genotyping
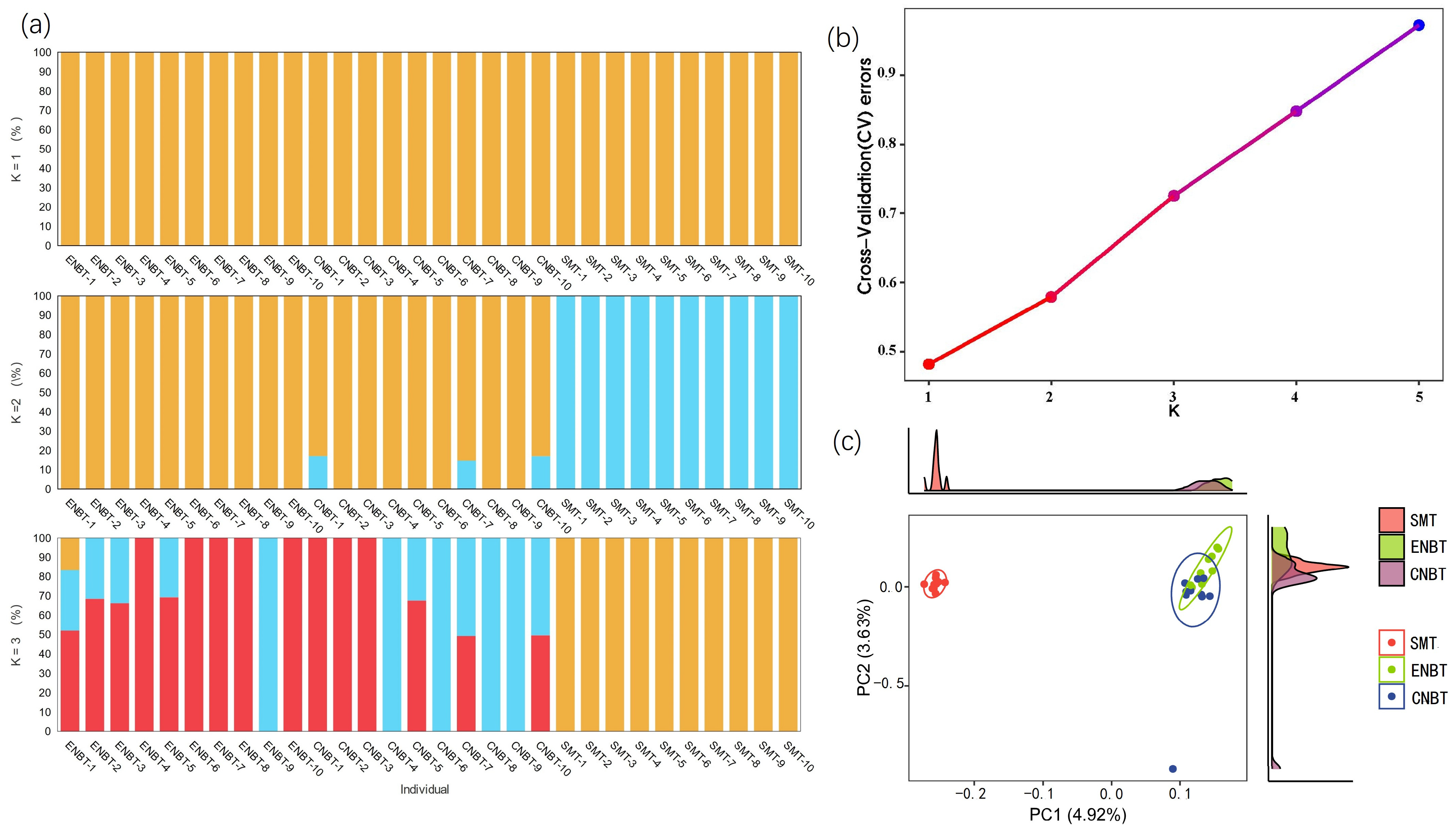
3.2. Genomic Inbreeding Level Estimation and Phylogenetic Analysis Regarding the Three Geographic Populations
3.3. Population Structure Analysis and Principal Component Analysis Regarding the A. gigantea Geographic Populations
3.4. Screening of the Environmental Adaptation Loci from the A. gigantea Geographic Populations
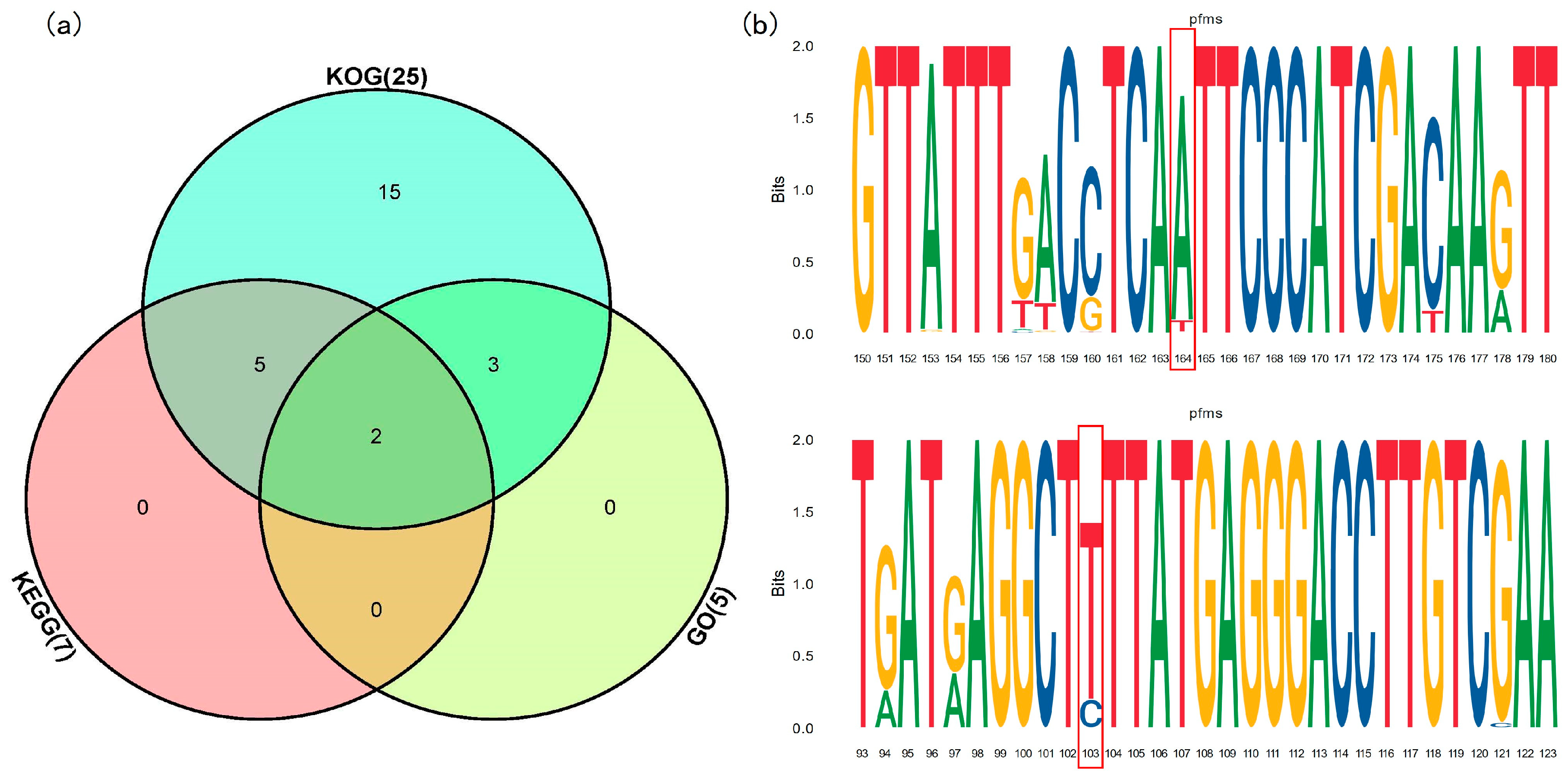
3.5. Functional Annotation of the Environmental Adaptation Loci
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, H.; Sun, C. Towards the understanding from sea surface to hadal zone—A multidisciplinary study of the Yap Trench. J. Oceanol. Limnol. 2020, 38, 591–592. [Google Scholar] [CrossRef]
- Cressey, D. The Hadal Zone: Life in the Deepest Oceans. Nature 2015, 523, SB1. [Google Scholar]
- Weston, J.N.J.; Espinosa-Leal, L.; Wainwright, J.A.; Stewart, E.C.D.; Gonzalez, C.E.; Linley, T.D.; Reid, W.D.K.; Hidalgo, P.; Oliva, M.E.; Ulloa, O.; et al. Eurythenes atacamensis sp. nov. (Crustacea: Amphipoda) exhibits ontogenetic vertical stratification across abyssal and hadal depths in the Atacama Trench, eastern South Pacific Ocean. Mar. Biodivers. 2021, 51, 51. [Google Scholar] [CrossRef]
- Chan, J.; Geng, D.; Pan, B.; Zhang, Q.; Xu, Q. Metagenomic Insights Into the Structure and Function of Intestinal Microbiota of the Hadal Amphipods. Front. Microbiol. 2021, 12, 668989. [Google Scholar] [CrossRef]
- Zhang, W.P.; Tian, R.M.; Sun, J.; Bougouffa, S.; Ding, W.; Cai, L.; Lan, Y.; Tong, H.Y.; Li, Y.X.; Jamieson, A.J.; et al. Genome Reduction in Psychromonas Species within the Gut of an Amphipod from the Ocean’s Deepest Point. Msystems 2018, 3, e00009-18. [Google Scholar] [CrossRef]
- Macdonald, A.G. Hydrostatic pressure as an environmental factor in life processes. Comp. Biochem. Phys. A 1997, 116, 291–297. [Google Scholar] [CrossRef]
- Huang, Y.; Sun, C.; Yang, G.; Yue, X.; Jiang, F.; Cao, W.; Yin, X.; Guo, C.; Niu, J.; Ding, H. Geochemical characteristics of hadal sediment in the northern Yap Trench. J. Oceanol. Limnol. 2019, 38, 650–664. [Google Scholar] [CrossRef]
- Eustace, R.M.; Ritchie, H.; Kilgallen, N.M.; Piertney, S.B.; Jamieson, A.J. Morphological and ontogenetic stratification of abyssal and hadal Eurythenes gryllus sensu lato (Amphipoda: Lysianassoidea) from the Peru-Chile Trench. Deep Sea Res. Part I 2016, 109, 91–98. [Google Scholar] [CrossRef]
- Lacey, N.C.; Rowden, A.A.; Clark, M.R.; Kilgallen, N.M.; Linley, T.; Mayor, D.J.; Jamieson, A.J. Community structure and diversity of scavenging amphipods from bathyal to hadal depths in three South Pacific Trenches. Deep Sea Res. Part I 2016, 111, 121–137. [Google Scholar] [CrossRef]
- Copilas-Ciocianu, D.; Borko, S.; Fiser, C. The late blooming amphipods: Global change promoted post-Jurassic ecological radiation despite Palaeozoic origin. Mol. Phylogenet. Evol. 2020, 143, 106664. [Google Scholar] [CrossRef]
- Shi, L.L.; Xiao, W.J.; Liu, Z.G.; Pan, B.B.; Xu, Y.P. Diet change of hadal amphipods revealed by fatty acid profile: A close relationship with surface ocean. Mar. Environ. Res. 2018, 142, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, F.; Jiang, S.; Pan, B.; Chan, J.; Xu, Q. The Adaptive Evolution and Gigantism Mechanisms of the Hadal “Supergiant” Amphipod Alicella gigantea. Front. Mar. Sci. 2021, 8, 743663. [Google Scholar] [CrossRef]
- Li, J.Y.; Song, Z.L.; Yan, G.Y.; He, L.S. The complete mitochondrial genome of the largest amphipod, Alicella gigantea: Insight into its phylogenetic relationships and deep sea adaptive characters. Int. J. Biol. Macromol. 2019, 141, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.W.; Davis, G.E. An Updated Classification of the Recent Crustacea; Natural History Museum of Los Angeles County: Los Angeles, CA, USA, 2001; 124p. [Google Scholar]
- Barnard, J.L.; Barnard, C.M. Freshwater Amphipoda of the World; Hayfield Associates: Mt. Vernon, VA, USA, 1983. [Google Scholar]
- Jamieson, A.J.; Lacey, N.C.; Lorz, A.N.; Rowden, A.A.; Piertney, S.B. The supergiant amphipod Alicella gigantea (Crustacea: Alicellidae) from hadal depths in the Kermadec Trench, SW Pacific Ocean. Deep Sea Res. Part II 2013, 92, 107–113. [Google Scholar] [CrossRef]
- Gallo, N.D.; Cameron, J.; Hardy, K.; Fryer, P.; Bartlett, D.H.; Levin, L.A. Submersible- and lander-observed community patterns in the Mariana and New Britain trenches: Influence of productivity and depth on epibenthic and scavenging communities. Deep Sea Res. Part I 2015, 99, 119–133. [Google Scholar] [CrossRef]
- Timofeev, S.F. Bergmann’s principle and deep-water gigantism in marine crustaceans. Biol. Bull. Russ. Acad. Sci. 2001, 28, 646–650. [Google Scholar] [CrossRef]
- Ritchie, H.; Jamieson, A.J.; Piertney, S.B. Genome size variation in deep-sea amphipods. R. Soc. Open Sci. 2017, 4, 170862. [Google Scholar] [CrossRef]
- Liu, Q.; Jiang, S.; Li, W.; Pan, B.; Xu, Q. Trimethylamine N-Oxide (TMAO) and Trimethylamine (TMA) Determinations of Two Hadal Amphipods. J. Mar. Sci. Eng. 2022, 10, 454. [Google Scholar] [CrossRef]
- Li, W.; Wang, F.; Jiang, S.; Pan, B.; Liu, Q.; Xu, Q. Morphological and molecular evolution of hadal amphipod’s eggs provides insights into embryogenesis under high hydrostatic pressure. Front. Cell Dev. Biol. 2022, 10, 987409. [Google Scholar] [CrossRef]
- Chan, J.L.; Geng, D.Q.; Pan, B.B.; Zhang, Q.M.; Xu, Q.H. Gut Microbial Divergence Between Three Hadal Amphipod Species from the Isolated Hadal Trenches. Microb. Ecol. 2022, 84, 627–637. [Google Scholar] [CrossRef]
- Zhu, L.; Geng, D.; Pan, B.; Li, W.; Jiang, S.; Xu, Q. Trace Elemental Analysis of the Exoskeleton, Leg Muscle, and Gut of Three Hadal Amphipods. Biol. Trace Elem. Res. 2022, 200, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Pan, B.; Geng, D.; Zhang, Q.; Zhang, S.; Guo, J.; Xu, Q. Genetic Diversity and Population Structure Analysis of Three Deep-Sea Amphipod Species from Geographically Isolated Hadal Trenches in the Pacific Ocean. Biochem. Genet. 2020, 58, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Yang, C.; Li, Q.; Zhu, W.; Chen, X.; Peng, M.; Chen, X.; Lin, Y.; Wang, H.; Liu, H.; et al. Identification of a quantitative trait loci (QTL) associated with ammonia tolerance in the Pacific white shrimp (Litopenaeus vannamei). BMC Genom. 2020, 21, 857. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xu, Y.; Gao, K.; Fan, G.; Zhang, F.; Deng, C.; Dai, S.; Huang, H.; Xin, H.; Li, Y. High-density genetic map construction and identification of loci controlling flower-type traits in Chrysanthemum (Chrysanthemum × morifolium Ramat.). Hortic. Res. 2020, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luan, P.; Ren, G.; Hu, G.; Yin, J. Estimating the inbreeding level and genetic relatedness in an isolated population of critically endangered Sichuan taimen (Hucho bleekeri) using genome-wide SNP markers. Ecol. Evol. 2020, 10, 1390–1400. [Google Scholar] [CrossRef]
- Fujioka, K.; Okino, K.; Kanamatsu, T.; Ohara, Y. Morphology and origin of the Challenger Deep in the Southern Mariana Trench. Geophys. Res. Lett. 2002, 29, 10-1–10-4. [Google Scholar] [CrossRef]
- Yancey, P.H. Cellular responses in marine animals to hydrostatic pressure. J. Exp. Zool. A Ecol. Integr. Physiol. 2020, 333, 398–420. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Qi, Y.Z.; Tian, J.W.; Wang, X.C.; Luo, C.L.; Zhou, C.; Zhang, X.H.; Xin, Y.; Wang, Y.H. Carbon cycling in the deep Mariana Trench in the western north Pacific Ocean: Insights from radiocarbon proxy data. Deep Sea Res. Part I 2020, 164, 103370. [Google Scholar] [CrossRef]
- Luo, M.; Gieskes, J.; Chen, L.Y.; Scholten, J.; Pan, B.B.; Lin, G.; Chen, D.F. Sources, Degradation, and Transport of Organic Matter in the New Britain Shelf-Trench Continuum, Papua New Guinea. J. Geophys. Res. Biogeosci. 2019, 124, 1680–1695. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microb. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Sun, B.; Lei, J. Specific-Locus Amplified Fragment Sequencing (SLAF-Seq) as High-Throughput SNP Genotyping Methods. Methods Mol. Biol. 2021, 2264, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Liang, Y.-K.; Liu, W.-C.; Chen, J.-H.; Chen, X.-R.; Tang, Q.; Sun, Z.-M.; Luan, M.-B. Relationships among Chinese Boehmeria species and the evolution of various clade. Ind. Crops Prod. 2020, 148, 112092. [Google Scholar] [CrossRef]
- Khan, S.A.; Chen, H.; Deng, Y.; Chen, Y.; Zhang, C.; Cai, T.; Ali, N.; Mamadou, G.; Xie, D.; Guo, B.; et al. High-density SNP map facilitates fine mapping of QTLs and candidate genes discovery for Aspergillus flavus resistance in peanut (Arachis hypogaea). Theor. Appl. Genet. 2020, 133, 2239–2257. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Du, M.; Xiong, M.; Chang, Y.; Liu, Z.; Wang, R.; Lin, X.; Zhou, Z.; Lu, M.; Liu, C.; Liu, E. Mining Candidate Genes and Favorable Haplotypes for Flag Leaf Shape in Rice (Oryza sativa L.) Based on a Genome-Wide Association Study. Agronomy 2022, 12, 1814. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Retief, J.D. Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 2000, 132, 243–258. [Google Scholar] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef]
- Coop, G.; Witonsky, D.; Di Rienzo, A.; Pritchard, J.K. Using Environmental Correlations to Identify Loci Underlying Local Adaptation. Genetics 2010, 185, 1411–1423. [Google Scholar] [CrossRef]
- Gunther, T.; Coop, G. Robust Identification of Local Adaptation from Allele Frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef]
- Li, Y.L.; Xue, D.X.; Zhang, B.D.; Liu, J.X. Population Genomic Signatures of Genetic Structure and Environmental Selection in the Catadromous Roughskin Sculpin Trachidermus fasciatus. Genome Biol. Evol. 2019, 11, 1751–1764. [Google Scholar] [CrossRef]
- Welcker, C.; Spencer, N.A.; Turc, O.; Granato, I.; Chapuis, R.; Madur, D.; Beauchene, K.; Gouesnard, B.; Draye, X.; Palaffre, C.; et al. Physiological adaptive traits are a potential allele reservoir for maize genetic progress under challenging conditions. Nat. Commun. 2022, 13, 3225. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Cooke, A.; Schwarzl, T.; Huppertz, I.; Kramer, G.; Mantas, P.; Alleaume, A.M.; Huber, W.; Krijgsveld, J.; Hentze, M.W. The RNA-Binding Protein YBX3 Controls Amino Acid Levels by Regulating SLC mRNA Abundance. Cell Rep. 2019, 27, 3097–3106. [Google Scholar] [CrossRef]
- Cano-Crespo, S.; Chillaron, J.; Junza, A.; Fernandez-Miranda, G.; Garcia, J.; Polte, C.; de la Ballina, L.R.; Ignatova, Z.; Yanes, O.; Zorzano, A.; et al. CD98hc (SLC3A2) sustains amino acid and nucleotide availability for cell cycle progression. Sci. Rep. 2019, 9, 14065. [Google Scholar] [CrossRef]
- Haddadi, N.; Lin, Y.G.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. “Dicing and Splicing” Sphingosine Kinase and Relevance to Cancer. Int. J. Mol. Sci. 2017, 18, 1891. [Google Scholar] [CrossRef] [PubMed]
- Weston, J.N.; Jensen, E.L.; Hasoon, M.S.; Kitson, J.J.; Stewart, H.A.; Jamieson, A.J. Barriers to gene flow in the deepest ocean ecosystems: Evidence from global population genomics of a cosmopolitan amphipod. Sci. Adv. 2022, 8, eabo6672. [Google Scholar] [CrossRef] [PubMed]
- France, S.C. Geographic variation among three isolated populations of the hadal amphipod Hirondellea gigas (Crustacea: Amphipoda: Lysianassoidea). Mar. Ecol.-Prog. Ser. 1993, 92, 277. [Google Scholar] [CrossRef]
- Qin, M.; Li, C.; Li, Z.; Chen, W.; Zeng, Y. Genetic Diversities and Differentially Selected Regions Between Shandong Indigenous Pig Breeds and Western Pig Breeds. Front. Genet. 2019, 10, 1351. [Google Scholar] [CrossRef] [PubMed]
- Rex, M.A.; Etter, R.J. Deep-Sea Biodiversity: Pattern and Scale; Harvard University Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Cowart, D.A.; Halanych, K.M.; Schaeffer, S.W.; Fisher, C.R. Depth-dependent gene flow in Gulf of Mexico cold seep Lamellibrachia tubeworms (Annelida, Siboglinidae). Hydrobiologia 2014, 736, 139–154. [Google Scholar] [CrossRef]
- Quattrini, A.M.; Baums, I.B.; Shank, T.M.; Morrison, C.L.; Cordes, E.E. Testing the depth-differentiation hypothesis in a deepwater octocoral. Proc. R. Soc. B Biol. Sci. 2015, 282, 20150008. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, H.; Cousins, N.; Cregeen, S.; Piertney, S. Population genetic structure of the abyssal grenadier (Coryphaenoides armatus) around the mid-Atlantic ridge. Deep. Sea Res. Part. II Top. Stud. Oceanogr. 2013, 98, 431–437. [Google Scholar] [CrossRef]
- Baco, A.R.; Etter, R.J.; Ribeiro, P.A.; Von der Heyden, S.; Beerli, P.; Kinlan, B.P. A synthesis of genetic connectivity in deep-sea fauna and implications for marine reserve design. Mol. Ecol. 2016, 25, 3276–3298. [Google Scholar] [CrossRef]
- Piertney, S.B.; Wenzel, M.; Jamieson, A.J. Large effective population size masks population genetic structure in Hirondellea amphipods within the deepest marine ecosystem, the Mariana Trench. Mol. Ecol. 2023, 32, 2206–2218. [Google Scholar] [CrossRef]
- Havermans, C.; Sonet, G.; d’Udekem d’Acoz, C.; Nagy, Z.T.; Martin, P.; Brix, S.; Riehl, T.; Agrawal, S.; Held, C. Genetic and morphological divergences in the cosmopolitan deep-sea amphipod Eurythenes gryllus reveal a diverse abyss and a bipolar species. PLoS ONE 2013, 8, e74218. [Google Scholar] [CrossRef] [PubMed]
- Palacin, M.; Errasti-Murugarren, E.; Rosell, A. Heteromeric amino acid transporters. In search of the molecular bases of transport cycle mechanisms. Biochem. Soc. Trans. 2016, 44, 745–752. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geographic Population | Sample Number | Geographic Coordinates | Depth (m) |
|---|---|---|---|
| ENBT 1 | 10 | S6.32°, E153.75° | 8931.3 |
| CNBT 2 | 10 | S5.86°, E152.43° | 8224.9 |
| SMT 3 | 10 | N11.62°, E142.35° | 6040 |
| Population | Sample Size | Genomic Inbreeding Coefficient (Mean ± SD) 1 | Min | Max |
|---|---|---|---|---|
| SMT | 10 | −0.123 a ± 0.041 | −0.1803 | −0.0525 |
| ENBT | 10 | −0.189 b ± 0.051 | −0.2881 | −0.1024 |
| CNBT | 10 | −0.113 a ± 0.091 | −0.1984 | 0.1304 |
| Total | 30 | −0.142 ± 0.071 | −0.2881 | 0.1304 |
| KEGG_B_Class | Pathway | Out (7) | All (44,233) |
|---|---|---|---|
| Immune system | Cytosolic DNA-sensing pathway | 3 | 3381 |
| Transcription | RNA polymerase | 3 | 3683 |
| Cell growth and death | Ferroptosis | 1 | 877 |
| Digestive system | Protein digestion and absorption | 1 | 1016 |
| Signal transduction | mTOR signaling pathway | 1 | 1035 |
| Signal transduction | VEGF signaling pathway | 1 | 1273 |
| Signal transduction | Sphingolipid signaling pathway | 1 | 1316 |
| Lipid metabolism | Sphingolipid metabolism | 1 | 1317 |
| Immune system | Fc gamma R-mediated phagocytosis | 1 | 1332 |
| Signal transduction | Apelin signaling pathway | 1 | 1408 |
| Signal transduction | Phospholipase D signaling pathway | 1 | 1597 |
| Signal transduction | Calcium signaling pathway | 1 | 1784 |
| Amino acid metabolism | Lysine degradation | 2 | 7024 |
| Infectious diseases | Tuberculosis | 1 | 2593 |
| Global and overview maps | Metabolic pathways | 3 | 17,672 |
| ID | Symbol | Pathway/Module |
|---|---|---|
| Marker63205 | SLC3A2, MDU1, CD98 | mTOR signaling pathway |
| Ferroptosis | ||
| Protein digestion and absorption | ||
| Marker137036 | SphK | Sphingosine degradation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Jiang, S.; Pan, B.; Xu, Q. Development of Single-Nucleotide Polymorphism Markers and Population Genetic Analysis of the Hadal Amphipod Alicella gigantea across the Mariana and New Britain Trenches. J. Mar. Sci. Eng. 2024, 12, 1117. https://doi.org/10.3390/jmse12071117
Chen L, Jiang S, Pan B, Xu Q. Development of Single-Nucleotide Polymorphism Markers and Population Genetic Analysis of the Hadal Amphipod Alicella gigantea across the Mariana and New Britain Trenches. Journal of Marine Science and Engineering. 2024; 12(7):1117. https://doi.org/10.3390/jmse12071117
Chicago/Turabian StyleChen, Lei, Shouwen Jiang, Binbin Pan, and Qianghua Xu. 2024. "Development of Single-Nucleotide Polymorphism Markers and Population Genetic Analysis of the Hadal Amphipod Alicella gigantea across the Mariana and New Britain Trenches" Journal of Marine Science and Engineering 12, no. 7: 1117. https://doi.org/10.3390/jmse12071117
APA StyleChen, L., Jiang, S., Pan, B., & Xu, Q. (2024). Development of Single-Nucleotide Polymorphism Markers and Population Genetic Analysis of the Hadal Amphipod Alicella gigantea across the Mariana and New Britain Trenches. Journal of Marine Science and Engineering, 12(7), 1117. https://doi.org/10.3390/jmse12071117