
Genetic Variability and Population Genetic Structure of Marbled Flounder Pseudopleuronectes yokohamae in Korea and Japan Inferred from mtDNA Control Region Sequences

Abstract
:1. Introduction
2. Materials and Methods
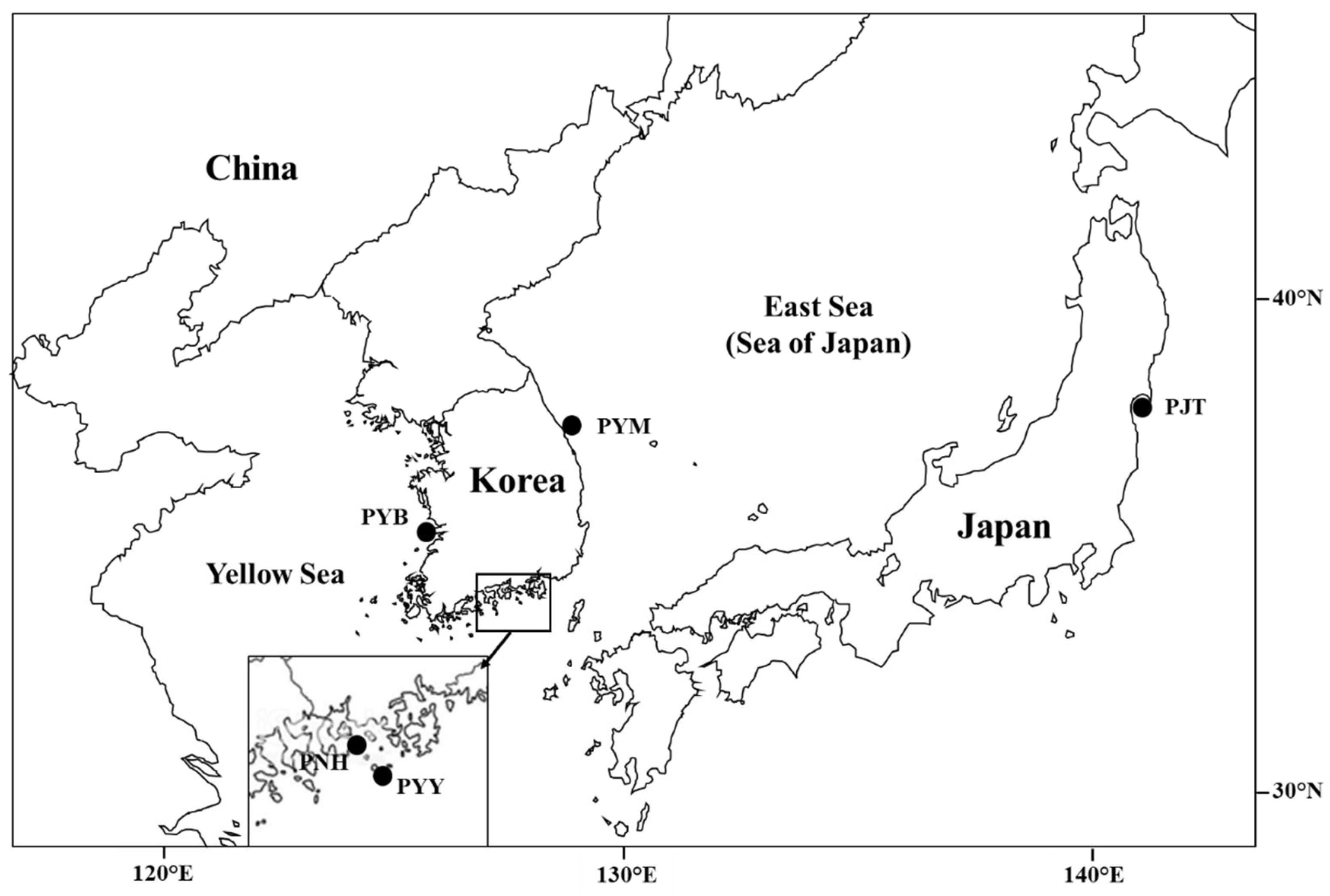
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Genetic Diversity and Population Genetic Analyses
3. Results
3.1. Genetic Diversity
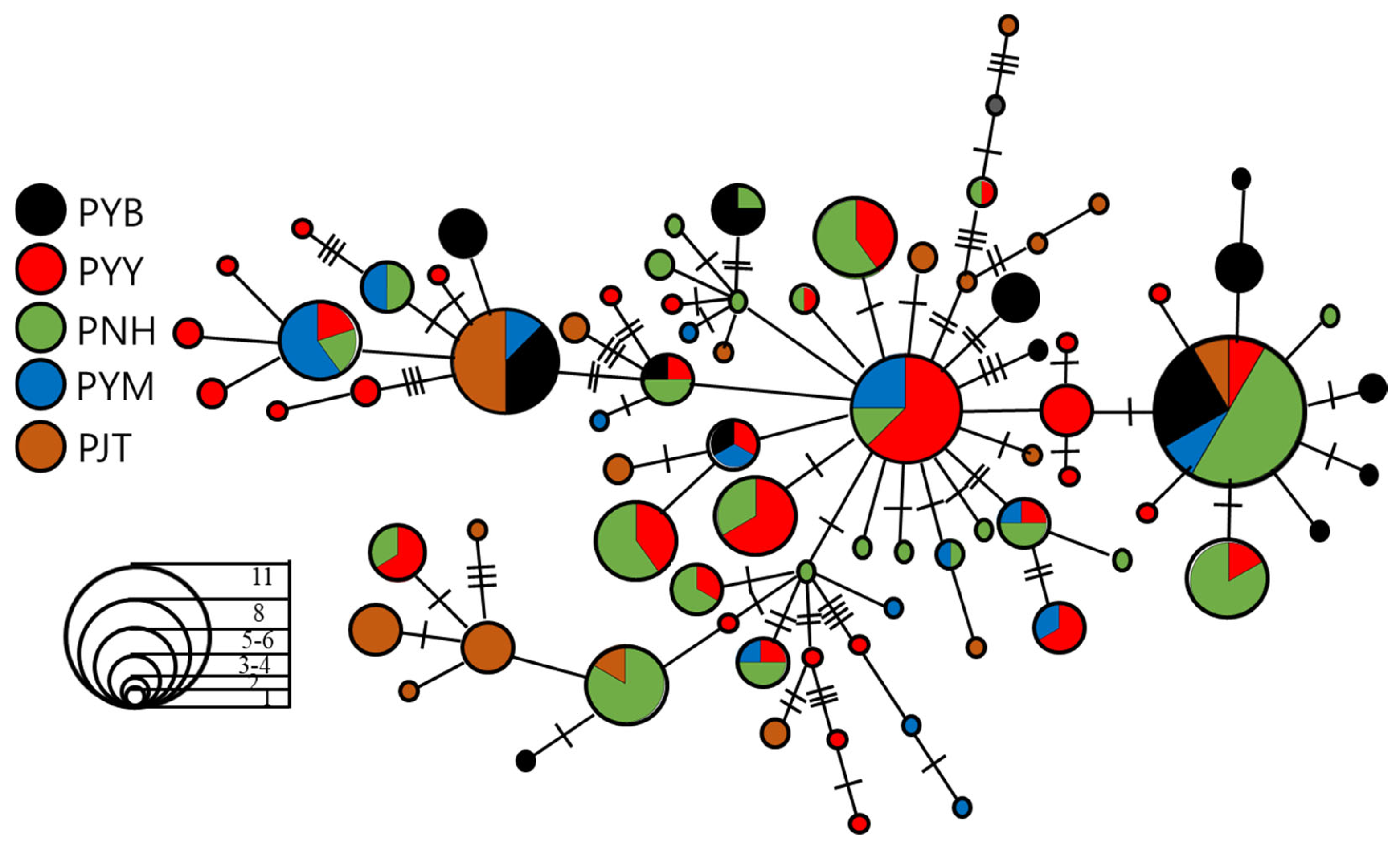
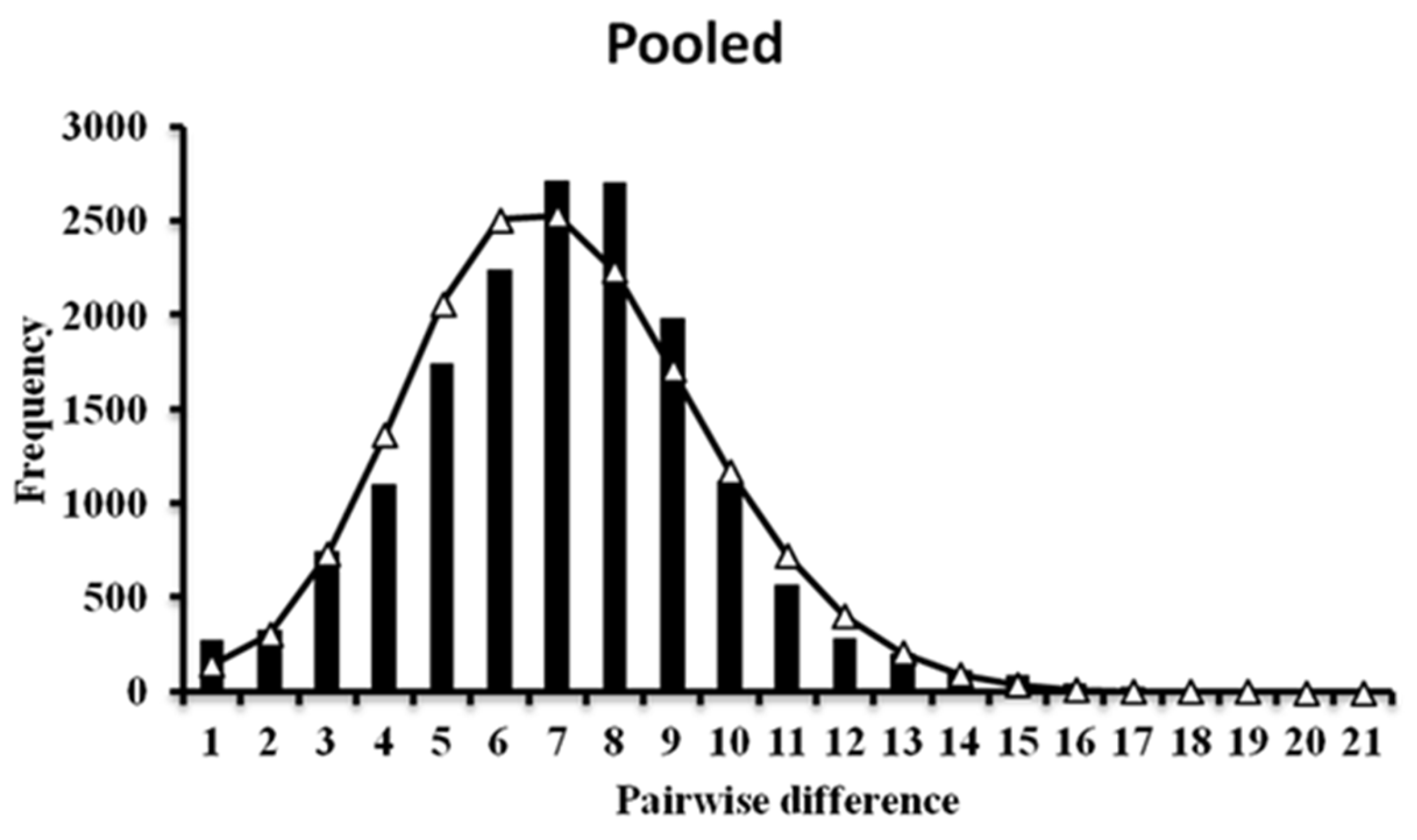
3.2. Demographic History and Population Structure
4. Discussion
4.1. Genetic Diversity and Demographic History
4.2. Population Genetic Structure
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, R.D.; Woodwark, M.; Skibinski, D.O.F. A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J. Fish Biol. 1994, 44, 213–232. [Google Scholar] [CrossRef]
- Gaylord, B.; Gaines, S.D. Temperature or transport? Range limits in marine species mediated solely by flow. Am. Nat. 2000, 155, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Schuller, M. Evidence for a role of bathymetry and emergence in speciation in the genus Glycera (Glyceridae, Polychaeta) from the deep Eastern Weddell Sea. Polar. Biol. 2011, 34, 549–564. [Google Scholar] [CrossRef]
- Marko, P.B.; Hart, M.W. The complex analytical landscape of gene flow inference. Am. Nat. 2011, 26, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.X. Response of Western Pacific marginal seas to glacial cycles: Aleoceangraphic and sedimentological features. Mar. Geol. 1999, 156, 5–39. [Google Scholar] [CrossRef]
- Nohara, K.; Takeuchi, H.; Tsuzaki, T.; Suzuki, N.; Tominaga, O.; Seikai, T. Genetic variability and stock structure of red tilefish Branchiostegus japonicus inferred from mitochondrial DNA sequence analysis. Fish. Sci. 2010, 76, 75–81. [Google Scholar] [CrossRef]
- Gwak, W.S.; Lee, Y.D.; Nakayama, K. Population structure and sequence divergence in the mitochondrial DNA control region of gizzard shad Konosirus punctatus in Korea and Japan. Ichthyol. Res. 2014, 62, 379–385. [Google Scholar] [CrossRef]
- Joh, M.; Takatsu, T.; Nakaya, M.; Higashitani, T.; Takahashi, T. Otolith microstructure and daily increment validation of marbled sole (Pseudopleuronectes yokohamae). Mar. Biol. 2005, 147, 59–69. [Google Scholar] [CrossRef]
- NFRDI (National Fisheries Research and Development Institute). Commercial Fishes of the Coastal & Offshore Waters in Korea; Yemunsa: Busan, Republic of Korea, 2004; 333p. [Google Scholar]
- Pearcy, W.G. Distribution and origin of demersal eggs within the order Pleuronectiformes. J. Cons. Int. Explor. Mer. 1962, 27, 233–235. [Google Scholar] [CrossRef]
- Joh, M.; Nakaya, M.; Yoshida, N.; Takatsu, T. Interannual growth differences and growth- selective survival in larvae and juveniles of marbled sole Pseudopleuronectes yokohamae. Mar. Ecol. Prog. Ser. 2013, 494, 267–279. [Google Scholar] [CrossRef]
- Jusan, K. Results of Mark-Recapture Experiments for Flatfish Species around Coastal Areas of Aomori Prefecture, Japan; Japan Fisheries Agency: Tokyo, Japan, 1988; pp. 4–12. (In Japanese) [Google Scholar]
- Joo, H.W.; Gwak, W.S. Estimation of early growth and spawning period of marbled flounder (Pseudopleuronectes yokohamae) in the water off Namhae of Korea as indicated from daily growth increments in otoliths. Korean Soc. Oceanogr. 2014, 19, 35–40. [Google Scholar]
- Kim, S.R.; Cha, H.K.; Lee, J.B.; Lee, H.W.; Yang, J.H.; Baek, H.J.; Kim, S.T. Maturity and spawning of the marbled flounder Pseudopleuronectes yokohamae off the coast of Pohang, East Sea. Korean J. Fish. Aquat. Sci. 2016, 49, 367–375. [Google Scholar] [CrossRef]
- Waples, R.S. Separating wheat from the chaff: Patterns of genetic differentiation in high gene flow species. J. Hered. 1998, 89, 438–450. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, S.G.; Gwak, W.S. Population genetic structure and genetic variability of the marbled sole Pleuronectes yokohamae on the coast of Gyeongsangnam-do, Korea. Anim. Cells. Syst. 2012, 16, 498–505. [Google Scholar] [CrossRef]
- Tsukagoshi, H.; Takeda, K.; Kariya, T.; Ozaki, T.; Takatsu, T.; Abe, S. Genetic variation and population structure of marbled sole Pleuronectes yokohamae and cresthead flounder P. schrenki in Japan inferred from mitochondrial DNA analysis. Biochem. Syst. Ecol. 2015, 58, 274–280. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Takanashi, A.; Yokosawa, Y.; Ikeda, M. Implication of homoplasy in hypervariable region (HVR) of mitochondrial DNA in a population of marbled flounder Pseudopleuronectes yokohamae: Consideration for conductingpopulation genetic analyses using the HVR. Fish. Sci. 2024, 1–12. [Google Scholar] [CrossRef]
- An, H.S.; Lee, J.W.; Park, J.Y.; Jung, H.T. Genetic structure of the Korean black scraper Thamnaconus modestus inferred from microsatellite marker analysis. Mol. Biol. Rep. 2013, 40, 3445–3456. [Google Scholar] [CrossRef]
- Avise, J.C. The history and preview of phytogeography: A personal reflection. Mol. Ecol. 1998, 7, 371–379. [Google Scholar] [CrossRef]
- Cann, R.L.; Brown, W.M.; Wilson, A.C. Polymorphic sites and the mechanism of evolution in human mitochondrial DNA. Genetics 1984, 106, 479–499. [Google Scholar] [CrossRef]
- Robalo, J.I.; Farias, I.; Francisco, S.M.; Avellaneda, K.; Castilho, R.; Figueiredo, I. Genetic population structure of Blackspot seabream (Pagellus bogarevio) contribution of mtDNA control region to fisheries management. Mitochondrial DNA Part A 2021, 32, 115–119. [Google Scholar] [CrossRef]
- Sun, C.H.; Yang, F.; Huang, Q.; Zeng, X.S.; Zhang, Y.N.; Li, S.; Yu, J.F.; Zhang, Q. Genetic population structure and demographic history of the endemic fish Paralichthys olivaceus of the Northwest Pacific Ocean. Echol. Evol. 2022, 12, e9506. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Saitou, N.; Nei, M. The neighbour-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony. Methods Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Teacher, A.G.F.; Griffiths, D.J. Hapstar: Automated haplotype network layout and visualization. Mol. Ecol. Resour. 2011, 11, 151–153. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic difference. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Bowen, B.W.; Muss, A.; Rocha, L.A.; Grant, W.S. Shallow mtDNA coalescence in Atlantic pygmy angelfishes (genus Centropyge) indicates a recent invasion from the Indian Ocean. J. Hered. 2006, 97, 1–12. [Google Scholar] [CrossRef]
- Xiao, Y.; Gao, T.; Zhang, Y.; Yanagimoto, T. Demographic history and population structureof Blackfin Flounder (Glyptocephalus stelleri) in Japan revealed by mitochondrial control region sequences. Biochem. Genet. 2010, 48, 402–417. [Google Scholar] [CrossRef]
- Sun, P.; Tang, B.J. Low mtDNA variation and shallow population structure of the Chinese pomfret Pampus chinensis along the China coast. J. Fish Biol. 2018, 92, 214–228. [Google Scholar] [CrossRef]
- Fujii, T.; Nishida, M. High sequence variability in the mitochondrial DNA control region of the Japanese flounder Paralichthys olivaceus. Fish. Sci. 1997, 63, 906–910. [Google Scholar] [CrossRef]
- Kim, S.G.; Morishima, K.; Arai, K. Genetic structure of wild brown sole inferred from mitochondrial DNA analysis. Anim. Cells Syst. 2010, 14, 197–206. [Google Scholar] [CrossRef]
- Grant, W.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Nielsen, E.E.; Hansen, J.H.; Larsen, P.F.; Bekkevold, D. Population genomics of marine fishes: Identifying adaptive variation in space and time. Mol. Ecol. 2009, 18, 3128–3150. [Google Scholar] [CrossRef]
- Imbrie, J.; Boyle, E.A.; Clemens, S.C.; Duvy, A.; Howard, W.R.; Kukla, G.; Kutzbach, J.; Martinson, D.G.; McIntyre, A.; Mix, A.C.; et al. On the structure and origin of major glaciation cycles, 1. Linear responses to Milankovitch forcing. Paleoceanography 1992, 7, 701–738. [Google Scholar] [CrossRef]
- Xu, X.; Oda, M. Surface-water evolution of the eastern East China Sea during the last 36,000 years. Mar. Geol. 1999, 156, 285–304. [Google Scholar] [CrossRef]
- Oba, T. Paleoenvironment of the Sea of Japan since the last glaciations. Chiku 1983, 5, 37–46. [Google Scholar]
- Liu, J.X.; Gao, T.X.; Zhuang, Z.M.; Jin, X.S.; Yokogawa, K.; Zhang, Y.P. Late Pleistocene divergence and subsequent population expansion of two closely related fish species, Japanese anchovy (Engraulis japonicus) and Australian anchovy (Engraulis australis). Mol. Phylogenet. Evol. 2006, 40, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Palumbi, S.R. Genetic divergence, Reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 1994, 25, 547–572. [Google Scholar] [CrossRef]
- Minegishi, Y.; Ikeda, M.; Kurita, Y.; Togashi, H.; Nakane, Y.; Kijima, A. Evaluation of the Tsunami impact on the genetic diversity of the marbled flounder Pseudopleuronectes yokohamae in Sendai Bay, Miyagi, Japan. Bull. Jpn. Fish. Res. Edu. Agen. 2017, 45, 69–73. [Google Scholar]
- Sato, M.; Kitanishia, S.; Ishiid, M.; Hamaguchia, M.; Kikuchie, K.; Hori, M. Genetic structure and demographic connectivity of marbled flounder (Pseudopleuronectes yokohamae) populations of Tokyo Bay. J. Sea Res. 2018, 142, 79–90. [Google Scholar] [CrossRef]
- Dou, S. Biology and ecology of flatfish in the Bohai Sea of China. Ph.D. Thesis, Chinese Academy of Sciences, Qingdao, China, 1994; pp. 1–139. [Google Scholar]
- Dou, S. Life history cycles of flatfish species in the bohai sea, China. Neth. J. Sea Res. 1995, 34, 195–210. [Google Scholar] [CrossRef]
- Dagang, C.; Changan, L.; Dou, S. The biology of flatfish (pleuronectinae) in the coastal waters of China. Neth. J. Sea Res. 1992, 29, 25–33. [Google Scholar] [CrossRef]
- Nakagami, M.; Takatsu, T.; Nakaya, M.; Takahashi, T. Spatial and temporal distribution of larval and juvenile marbled sole Pleuronectes yokohamae in Hakodate Bay. Bull. Jpn. Soc. Fish. Oceanogr. 2001, 65, 85–93, (In Japanese with English abstract). [Google Scholar]
- Lee, J.H.; Kodama, K.; Oyama, M.; Shiraishi, H.; Horiguchi, T. Effect of water temperature on survival of early-life stages of marbled flounder Pseudopleuronectes yokohamae in Tokyo Bay, Japan. Mar. Environ. Res. 2016, 128, 107–113. [Google Scholar] [CrossRef]
- Suda, A.; Nagata, N.; Sato, A.; Narimatsu, Y.; Nadiatul, H.H.; Kawata, M. Genetic variation and local differences in Pacific cod Gadus macrocephalus around Japan. J. Fish Biol. 2017, 90, 61–79. [Google Scholar] [CrossRef]
- Yun, J.Y.; Magaard, L.; Kim, K.; Shin, C.W.; Kim, C.; Byun, S.K. Spatial and temporal variability of the North Korean cold water leading to the near-bottom cold water intrusion in Korea Strait. Progr. Oceanogr. 2004, 60, 99–131. [Google Scholar] [CrossRef]
- Chen, C.T.A. Chemical and physical fronts in the Bohai, Yellow and East China seas. J. Mar. Syst. 2009, 78, 394–410. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, H.Q.; Fang, H.G. A numerical study on the path and origin of Yellow Sea warm current. Yellow Sea 1997, 3, 18–26. [Google Scholar]
- Kim, W.J.; Kim, K.K.; Han, H.S.; Nam, B.H.; Kim, Y.O.; Kong, H.J.; Noh, J.K.; Yoon, M. Population structure of the olive flounder (Paralichthys olivaceus) in Korea inferred from microsatellite marker analysis. J. Fish Biol. 2010, 76, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Itaki, T.; Minoshima, K.; Hodaka, K. Radiolarian flux at an IMAGES site at the western margin of the subarctic Pacific and its seasonal relationship to the Oyashio Cold and Tsugaru Warm currents. Mar. Geol. 2008, 255, 131–148. [Google Scholar] [CrossRef]
- Verma, R.; Singh, M.; Kumar, S. Unraveling the limits of mitochondrial control region to estimate the fine scale population genetic differentiation in anadromous fish Tenualosa ilisha. Scientifica 2016, 2016, 2035240. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Coast | Sampling Site (Abbr.) | n | Date of Collection | Source |
|---|---|---|---|---|---|
| Korea | West | Biungdo (PYB) | 32 | April 2016 | Present study |
| South | Yokjido (PYY) | 52 | February 2010, February 2011 | Lee et al. (2012) [16] | |
| Namhae (PNH) | 51 | January 2011, February 2011, March 2011 | Lee et al. (2012) [16] | ||
| East | Mukho (PYM) | 17 | February 2012 | Present study | |
| Japan | Tohoku (PJT) | 29 | February 2014 | Present study |
| Location | n | ts + tv | S | N | h (SD) | π (SD) |
|---|---|---|---|---|---|---|
| PYB | 32 | 20 + 5 | 25 | 15 | 0.944 ± 0.017 | 0.015 ± 0.008 |
| PYY | 52 | 42 + 3 | 45 | 36 | 0.980 ± 0.008 | 0.016 ± 0.008 |
| PNH | 51 | 35 + 3 | 38 | 27 | 0.964 ± 0.010 | 0.014 ± 0.007 |
| PYM | 17 | 26 + 3 | 29 | 15 | 0.986 ± 0.025 | 0.017 ± 0.009 |
| PJT | 29 | 34 + 3 | 37 | 18 | 0.961 ± 0.018 | 0.017 ± 0.009 |
| All | 181 | 60 + 10 | 70 | 81 | 0.983 ± 0.003 | 0.016 ± 0.008 |
| Location | Haplotype | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PYY01 | PYY02 | PYY48 | PYM17 | PYY05 | PYM16 | PNH52 | PNH43 | PNH35 | PYM13 | PYY12 | PNH19 | PNH29 | |
| PYB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYY | 1 | 1 | 2 | 5 | 1 | 1 | 2 | 4 | 1 | 2 | 1 | 2 | 1 |
| PNH | 0 | 0 | 0 | 1 | 0 | 2 | 3 | 2 | 1 | 0 | 0 | 1 | 2 |
| PYM | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| PJT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYB11 | PNH48 | PYB28 | PYY24 | PNH42 | PYY54 | PYY27 | PYB26 | PYY30 | PYY31 | PYY33 | PYY41 | PYY35 | |
| PYB | 1 | 0 | 4 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| PYY | 1 | 2 | 1 | 1 | 1 | 3 | 1 | 1 | 1 | 1 | 1 | 2 | 1 |
| PNH | 2 | 3 | 1 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| PJT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYY37 | PYY38 | PYY39 | PYY43 | PYY44 | PNH53 | PNH51 | PYY47 | PYY51 | PYB20 | PNH01 | PJT12 | PNH08 | |
| PYB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 |
| PYY | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 |
| PNH | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | 5 | 1 | 5 | 1 |
| PYM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| PJT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 |
| PNH12 | PNH13 | PYB27 | PYM03 | PNH22 | PYM10 | PNH39 | PNH40 | PNH47 | PYM20 | PYM02 | PYM04 | PYM06 | |
| PYB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYY | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PNH | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | 0 | 0 |
| PYM | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
| PJT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYM08 | PJT29 | PYM22 | PYM29 | PYB05 | PYB10 | PYB30 | PYB18 | PYB19 | PYB21 | PYB32 | PYB24 | PYB31 | |
| PYB | 0 | 4 | 0 | 0 | 0 | 0 | 3 | 3 | 2 | 1 | 3 | 1 | 1 |
| PYY | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PNH | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYM | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PJT | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PJT09 | PJT28 | PJT08 | PJT20 | PJT24 | PJT10 | PJT21 | PJT15 | PJT16 | PJT17 | PJT18 | PJT23 | PJT26 | |
| PYB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYY | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PNH | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PYM | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| PJT | 2 | 3 | 2 | 2 | 3 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 |
| PJT27 | PJT31 | PJT32 | |||||||||||
| PYB | 0 | 0 | 0 | ||||||||||
| PYY | 0 | 0 | 0 | ||||||||||
| PNH | 0 | 0 | 0 | ||||||||||
| PYM | 0 | 0 | 0 | ||||||||||
| PJT | 1 | 1 | 1 | ||||||||||
| Population Expansion Parameters | Mismatch Distribution Parameters | ||||
|---|---|---|---|---|---|
| Location | Tajima’s D | Fu’s FS | τ | SSD | Hri |
| PYB | −0.484 | −2.398 * | 7.193 | 0.011 | 0.020 |
| PYY | −1.255 | −24.751 * | 6.875 | 0.005 | 0.016 |
| PNH | −1.256 | −12.561 * | 5.742 | 0.003 | 0.017 |
| PYM | −0.966 | −6.999 * | 3.980 | 0.047 | 0.029 |
| PJT | −0.857 | −4.917 * | 7.015 | 0.005 | 0.017 |
| All | −1.423 * | −24.901 * | 6.414 | 0.001 | 0.410 |
| Location | PYB | PYY | PNH | PYM | PJT |
|---|---|---|---|---|---|
| PYB | 0.006 | 0.000 | 0.012 | 0.002 | |
| PYY | 0.038 | 0.102 | 0.480 | 0.001 | |
| PNH | 0.069 | 0.009 | 0.113 | 0.000 | |
| PYM | 0.066 | −0.002 | 0.018 | 0.093 | |
| PJT | 0.075 | 0.038 | 0.066 | 0.024 |
| Comparisons | Source of Variation | Sum of Squares | % Variation | Fixation Indices | p |
|---|---|---|---|---|---|
| One-group (PYB, PYY, PNH, PYM, PJT) | Among populations | 28.698 | 4.60 | 0.049 | 0.000 |
| Within populations | 521.992 | 95.40 | |||
| Total | 550.691 | 100 | |||
| Three-group (PYB) (PYY, PNH, PYM) (PJT) | Among groups | 20.999 | 4.71 | 0.011 | 0.00 |
| Among populations Within groups | 7.699 | 0.77 | 0.054 | 0.10 | |
| Within populations | 521.992 | 94.52 | 0.047 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gwak, W.-S.; Roy, A. Genetic Variability and Population Genetic Structure of Marbled Flounder Pseudopleuronectes yokohamae in Korea and Japan Inferred from mtDNA Control Region Sequences. J. Mar. Sci. Eng. 2024, 12, 1506. https://doi.org/10.3390/jmse12091506
Gwak W-S, Roy A. Genetic Variability and Population Genetic Structure of Marbled Flounder Pseudopleuronectes yokohamae in Korea and Japan Inferred from mtDNA Control Region Sequences. Journal of Marine Science and Engineering. 2024; 12(9):1506. https://doi.org/10.3390/jmse12091506
Chicago/Turabian StyleGwak, Woo-Seok, and Animesh Roy. 2024. "Genetic Variability and Population Genetic Structure of Marbled Flounder Pseudopleuronectes yokohamae in Korea and Japan Inferred from mtDNA Control Region Sequences" Journal of Marine Science and Engineering 12, no. 9: 1506. https://doi.org/10.3390/jmse12091506