
Genome-Wide Marker Analysis for Traits of Economic Importance in Asian Seabass Lates calcarifer



Abstract
:1. Introduction
2. Materials and Methods
2.1. Fish Samples
2.2. Traits Studied
2.3. DNA Extraction, Preparation of Libraries, and Sequencing
2.4. Genome-Wide Association Study (GWAS)
2.5. Genomic Prediction
3. Results
3.1. Phenotypic Data
3.2. Heritability and Correlation
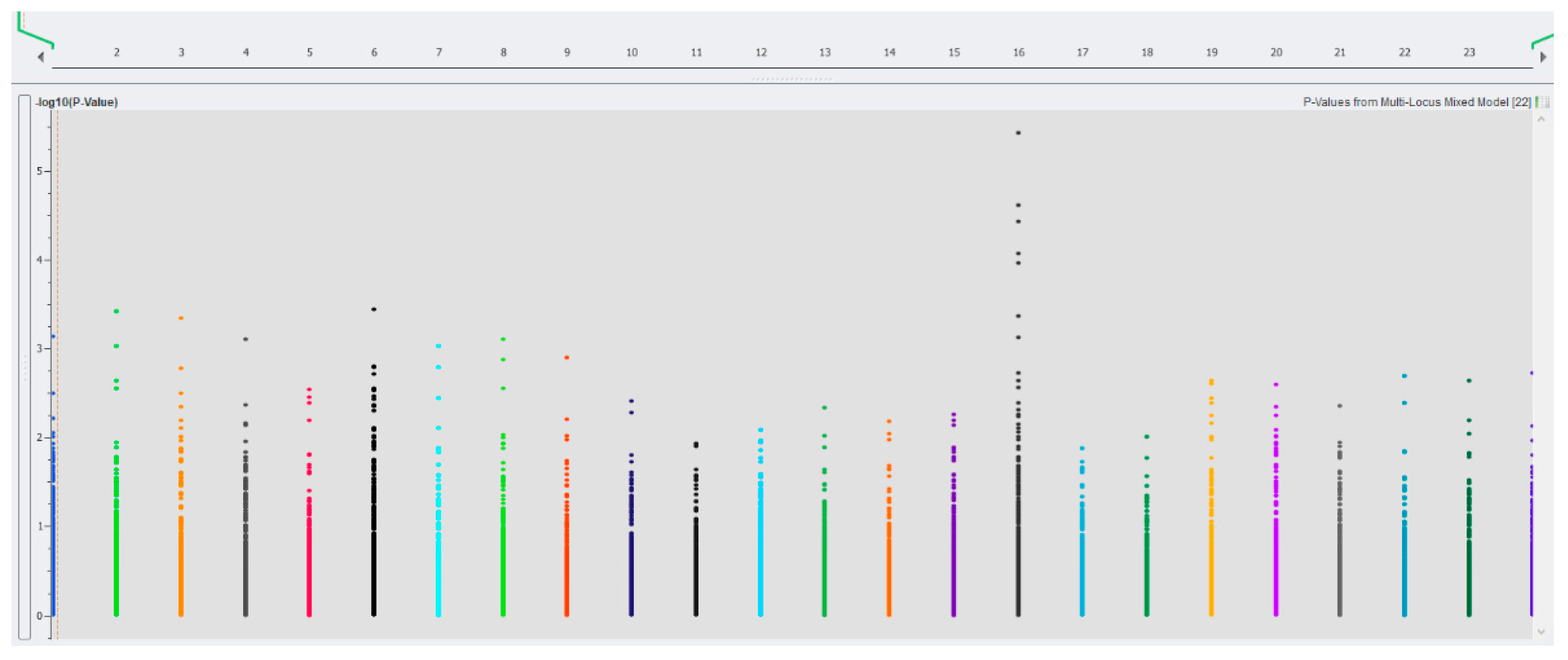
3.3. SNPs Associated With Traits of Economic Importance
3.4. Accuracy of Genomic Prediction
4. Discussion
4.1. Genomic Prediction
4.2. Significant Markers
4.3. Genetic Parameters
4.4. Practical Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Khang, P.V.; Phuong, T.H.; Dat, N.K.; Knibb, W.; Nguyen, N.H. An 8-Year Breeding Program for Asian Seabass Lates calcarifer: Genetic Evaluation, Experiences, and Challenges. Front. Genet. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed]
- Khang, P.V.; Nha, V.V.; Nguyen, N.H. Resistance to Streptococcus iniae and its genetic associations with traits of economic importance in Asian seabass (Lates calcarifer). J. Fish Dis. 2019, 42, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Hamzah, A.; Thoa, N.P. Effects of genotype by environment interaction on genetic gain and genetic parameter estimates in Red Tilapia (Oreochromis spp.). Front. Genet. 2017, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Hung, D.; Vu, N.T.; Nguyen, N.H.; Ponzoni, R.W.; Hurwood, D.A.; Mather, P.B. Genetic response to combined family selection for improved mean harvest weight in giant freshwater prawn (Macrobrachium rosenbergii) in Vietnam. Aquaculture 2013, 412, 70–73. [Google Scholar] [CrossRef]
- In, V.V.; Sang, V.V.; O’Connor, W.; Van, P.T.; Dove, M.; Knibb, W.; Nguyen, N.H. Are strain genetic effect and heterosis expression altered with culture system and rearing environment in the Portuguese oyster (Crassostrea angulata)? Aquac. Res. 2017, 48, 4058–4069. [Google Scholar] [CrossRef]
- Xia, J.H.; Lin, G.; He, X.; Yunping, B.; Liu, P.; Liu, F.; Sun, F.; Tu, R.; Yue, G.H. Mapping quantitative trait loci for omega-3 fatty acids in Asian seabass. Mar. Biotechnol. 2014, 16, 1–9. [Google Scholar] [CrossRef]
- Wang, L.; Bai, B.; Huang, S.; Liu, P.; Wan, Z.Y.; Ye, B.; Wu, J.; Yue, G.H. QTL mapping for resistance to iridovirus in Asian seabass using genotyping-by-sequencing. Mar. Biotechnol. 2017, 19, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, L.; Wong, S.-M.; Yue, G.H. Fine mapping QTL for resistance to VNN disease using a high-density linkage map in Asian seabass. Sci. Rep. 2016, 6, 32122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.H.; Lin, G.; He, X.; Liu, P.; Liu, F.; Sun, F.; Tu, R.; Yue, G.H. Whole genome scanning and association mapping identified a significant association between growth and a SNP in the IFABP-a gene of the Asian seabass. BMC Genom. 2013, 14, 295. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, P.; Huang, S.; Ye, B.; Chua, E.; Wan, Z.Y.; Yue, G.H. Genome-wide association study identifies loci associated with resistance to viral nervous necrosis disease in Asian seabass. Mar. Biotechnol. 2017, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xia, J.; Wang, C.; Pang, H.; Yue, G. Significant associations of polymorphisms in the prolactin gene with growth traits in Asian seabass (Lates calcarifer). Anim. Genet. 2012, 43, 233–236. [Google Scholar] [CrossRef]
- Fu, G.H.; Bai, Z.Y.; Xia, J.H.; Liu, X.J.; Liu, F.; Wan, Z.Y.; Yue, G.H. Characterization of the LECT2 gene and its associations with resistance to the big belly disease in Asian seabass. Fish Shellfish Immunol. 2014, 37, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Babu, P.G.; Jeena, K.; Tripathi, G.; Prasad, K.P. Molecular characterization, ontogeny and expression profiling of mitochondrial antiviral signaling adapter, MAVS from Asian seabass Lates calcarifer, Bloch (1790). Dev. Comp. Immunol. 2018, 79, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.K.; Truong, H.P.; Nguyen, D.K.; Nguyen, N.H. Genetic component of cannibalism in Asian seabass Lates Calcarifer. Appl. Anim. Behav. Sci. 2020, 231. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Premachandra, H.; Kilian, A.; Knibb, W. Genomic prediction using DArT-Seq technology for yellowtail kingfish Seriola lalandi. BMC Genom. 2018, 19, 107. [Google Scholar] [CrossRef]
- Vij, S.; Kuhl, H.; Kuznetsova, I.S.; Komissarov, A.; Yurchenko, A.A.; Van Heusden, P.; Singh, S.; Thevasagayam, N.M.; Prakki, S.R.S.; Purushothaman, K.J.P.g. Chromosomal-level assembly of the Asian seabass genome using long sequence reads and multi-layered scaffolding. PLoS Genet. 2016, 12, e1005954. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.H.; Rastas, P.M.; Premachandra, H.; Knibb, W. First high-density linkage map and single nucleotide polymorphisms significantly associated with traits of economic importance in Yellowtail Kingfish Seriola lalandi. Front. Genet. 2018, 9, 127. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- SNP & Variation Suite™ (Version 8.9.0) [Software]. Golden Helix, Inc.: Bozeman, MT, USA. Available online: http://www.goldenhelix.com (accessed on 5 March 2021).
- Tsai, H.-Y.; Hamilton, A.; Tinch, A.E.; Guy, D.R.; Gharbi, K.; Stear, M.J.; Matika, O.; Bishop, S.C.; Houston, R.D. Genome wide association and genomic prediction for growth traits in juvenile farmed Atlantic salmon using a high density SNP array. BMC Genom. 2015, 16, 969. [Google Scholar] [CrossRef] [Green Version]
- Robledo, D.; Matika, O.; Hamilton, A.; Houston, R.D. Genome-wide association and genomic selection for resistance to amoebic gill disease in Atlantic salmon. G3 Genes Genomes Genet. 2018, 8, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Barria, A.; Christensen, K.A.; Yoshida, G.M.; Correa, K.; Jedlicki, A.; Lhorente, J.P.; Davidson, W.S.; Yáñez, J.M. Genomic predictions and genome-wide association study of resistance against Piscirickettsia salmonis in coho salmon (Oncorhynchus kisutch) using ddRAD sequencing. G3 Genes Genomes Genet. 2018, 8, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.M.; Carvalheiro, R.; Rodríguez, F.H.; Lhorente, J.P.; Yáñez, J.M. Single-step genomic evaluation improves accuracy of breeding value predictions for resistance to infectious pancreatic necrosis virus in rainbow trout. Genomics 2019, 111, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Palaiokostas, C.; Cariou, S.; Bestin, A.; Bruant, J.-S.; Haffray, P.; Morin, T.; Cabon, J.; Allal, F.; Vandeputte, M.; Houston, R.D. Genome-wide association and genomic prediction of resistance to viral nervous necrosis in European sea bass (Dicentrarchus labrax) using RAD sequencing. Genet. Sel. Evol. 2018, 50, 30. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.L.; Carraro, R.; Bestin, A.; Cariou, S.; Sonesson, A.K.; Bruant, J.-S.; Haffray, P.; Bargelloni, L.; Meuwissen, T.H.E. Genetics of resistance to photobacteriosis in gilthead sea bream (Sparus aurata) using 2b-RAD sequencing. BMC Genet. 2018, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Phuthaworn, C.; Knibb, W. Genomic prediction for disease resistance to Hepatopancreatic parvovirus and growth, carcass and quality traits in Banana shrimp Fenneropenaeus merguiensis. Genomics 2020, 112, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wan, Z.Y.; Bai, B.; Huang, S.Q.; Chua, E.; Lee, M.; Pang, H.Y.; Wen, Y.F.; Liu, P.; Liu, F. Construction of a high-density linkage map and fine mapping of QTL for growth in Asian seabass. Sci. Rep. 2015, 5, 16358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neto, R.V.R.; Yoshida, G.M.; Lhorente, J.P.; Yáñez, J.M. Genome-wide association analysis for body weight identifies candidate genes related to development and metabolism in rainbow trout (Oncorhynchus mykiss). Mol. Genet. Genom. 2019, 294, 1–9. [Google Scholar]
- Holborn, M.K.; Ang, K.P.; Elliott, J.; Powell, F.; Boulding, E.G. Genome wide association analysis for bacterial kidney disease resistance in a commercial North American Atlantic salmon (Salmo salar) population using a 50 K SNP panel. Aquaculture 2018, 495, 465–471. [Google Scholar] [CrossRef]
- Robledo, D.; Gutierrez, A.P.; Barria, A.; Lhorente, J.P.; Houston, R.D.; Yáñez, J.M. Discovery and functional annotation of quantitative trait loci affecting resistance to sea lice in Atlantic salmon. Front. Genet. 2019, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A. Finding the missing heritability of complex diseases. Nature 2009, 461, 747. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Trait | Unit | n | Mean | SD | CV | SNP Heritability | Accuracy |
|---|---|---|---|---|---|---|---|
| Weight | g | 187 | 281.0 | 131.2 | 46.7 | 0.26 ± 0.15 | 0.78 |
| Length | cm | 187 | 27.5 | 4.3 | 15.8 | 0.07 ± 0.11 | 0.67 |
| Survival | % | 184 | 57.5 | 13.9 | 32.8 | 0.01 ± 0.07 | 0.25 |
| Cannibalism | % | 184 | 30.6 | 10.1 | 33.0 | 0.05 × 10−3 ± 0.09 | n.e. |
| Disease resistance | % | 184 | 57.5 | 13.9 | 24.2 | 0.01 ± 0.08 | n.e. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.H.; Van Khang, P. Genome-Wide Marker Analysis for Traits of Economic Importance in Asian Seabass Lates calcarifer. J. Mar. Sci. Eng. 2021, 9, 282. https://doi.org/10.3390/jmse9030282
Nguyen NH, Van Khang P. Genome-Wide Marker Analysis for Traits of Economic Importance in Asian Seabass Lates calcarifer. Journal of Marine Science and Engineering. 2021; 9(3):282. https://doi.org/10.3390/jmse9030282
Chicago/Turabian StyleNguyen, Nguyen Hong, and Pham Van Khang. 2021. "Genome-Wide Marker Analysis for Traits of Economic Importance in Asian Seabass Lates calcarifer" Journal of Marine Science and Engineering 9, no. 3: 282. https://doi.org/10.3390/jmse9030282
APA StyleNguyen, N. H., & Van Khang, P. (2021). Genome-Wide Marker Analysis for Traits of Economic Importance in Asian Seabass Lates calcarifer. Journal of Marine Science and Engineering, 9(3), 282. https://doi.org/10.3390/jmse9030282