
Structural and Functional Modeling of Artificial Bioactive Proteins

Abstract
:1. Introduction
2. Results and Discussion
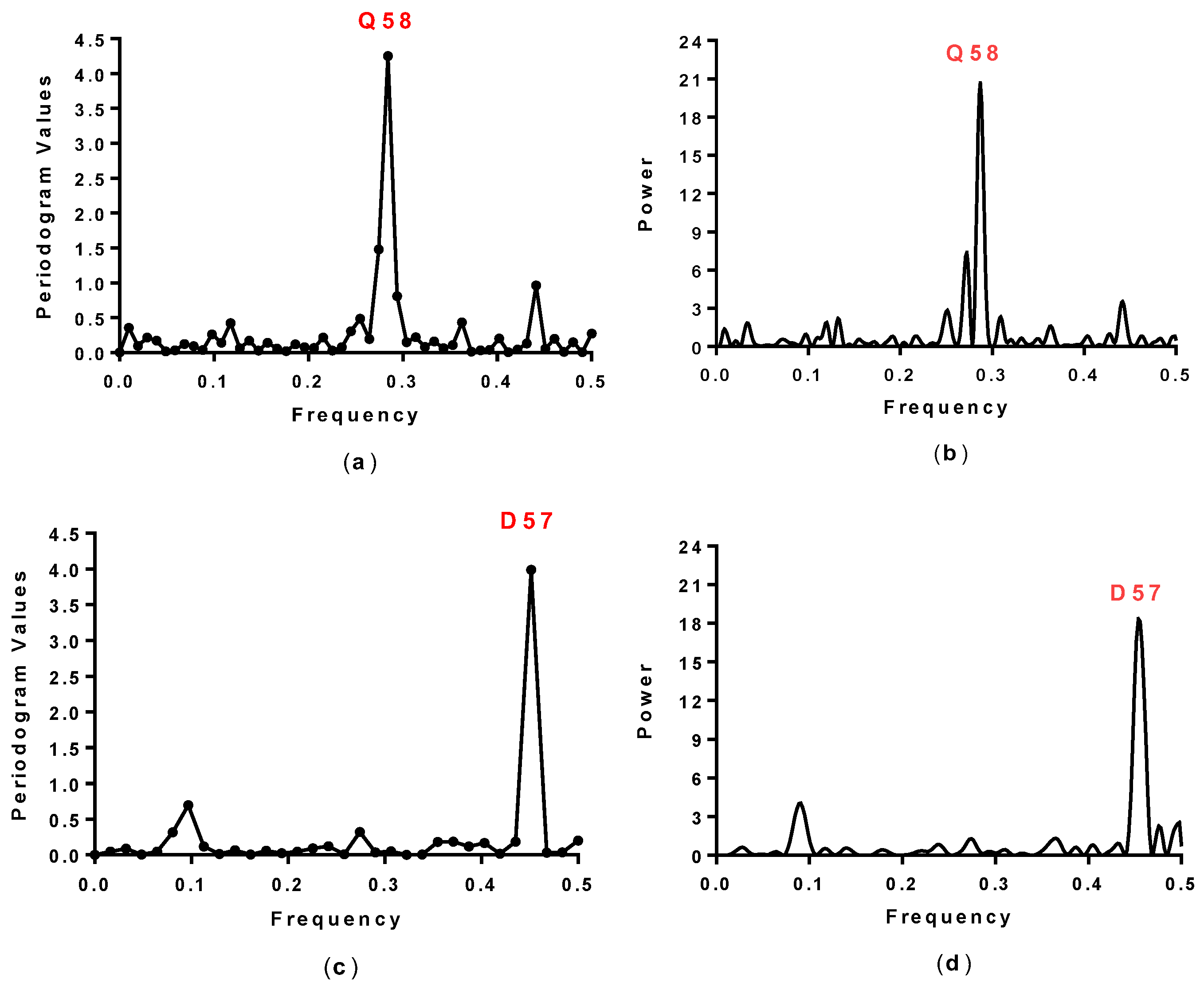
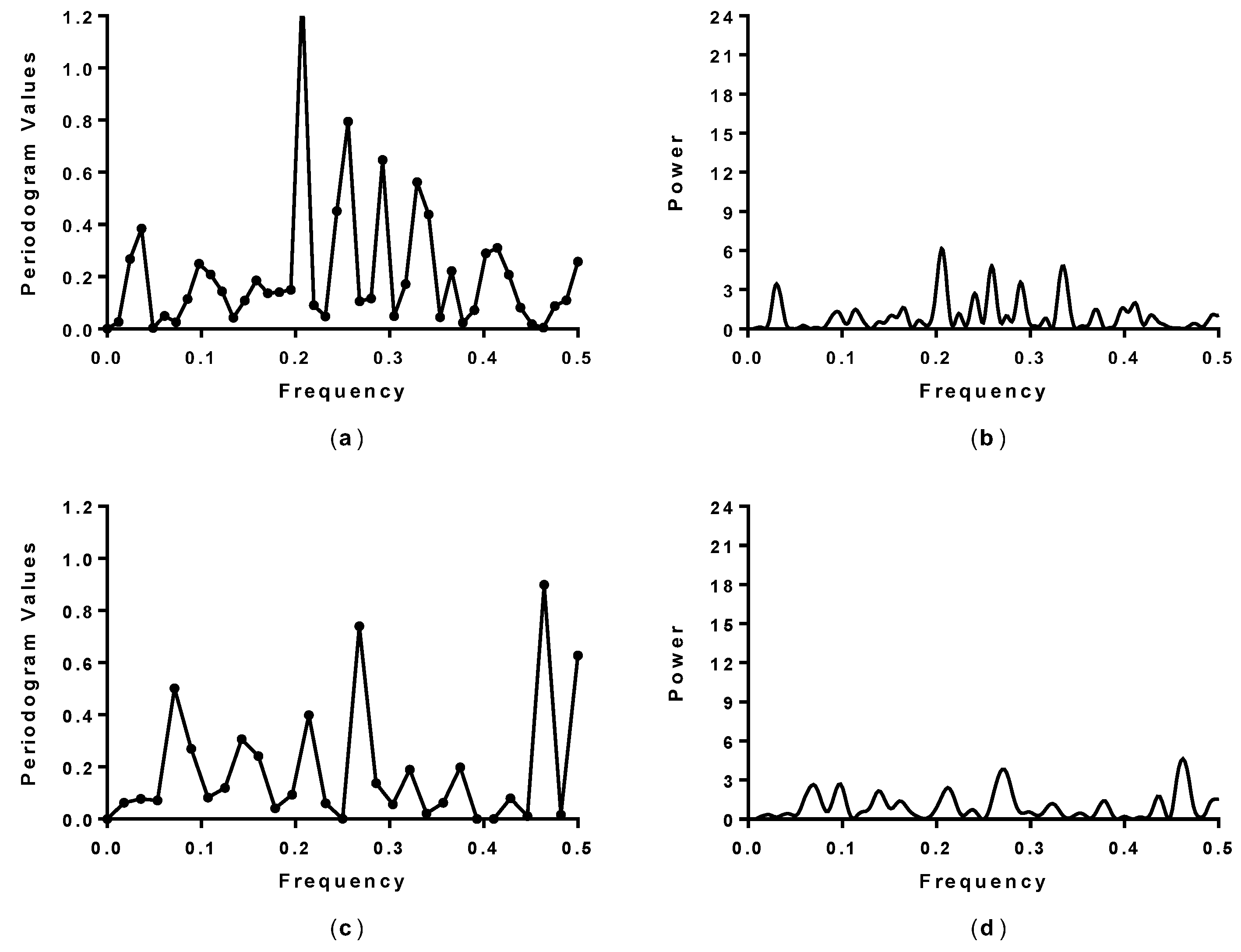
2.1. Spectral Analysis of Hecht_α and Hecht_β Proteins
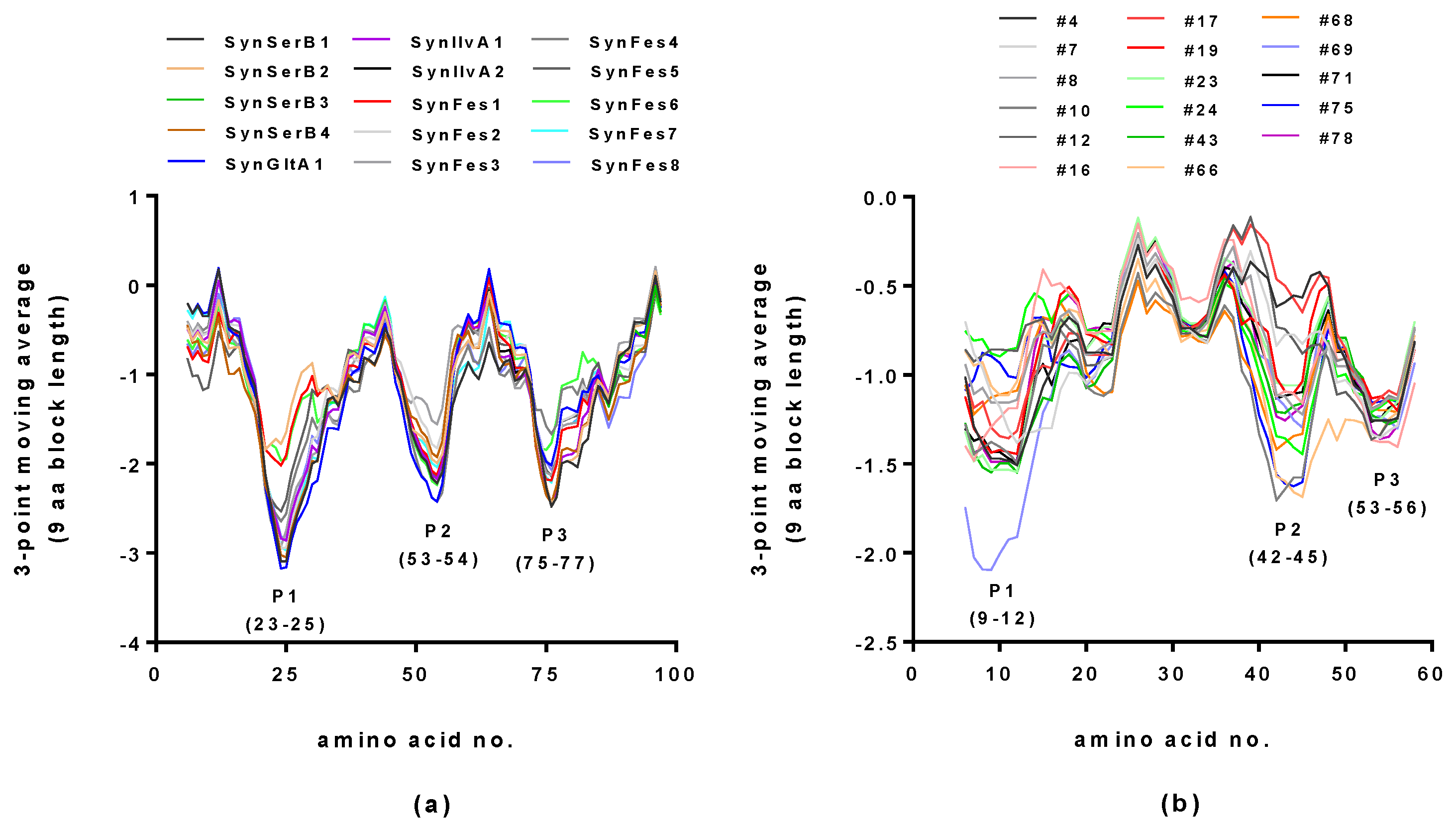
2.2. Hydropathy Analyses of Hecht_α and Hecht_β Proteins
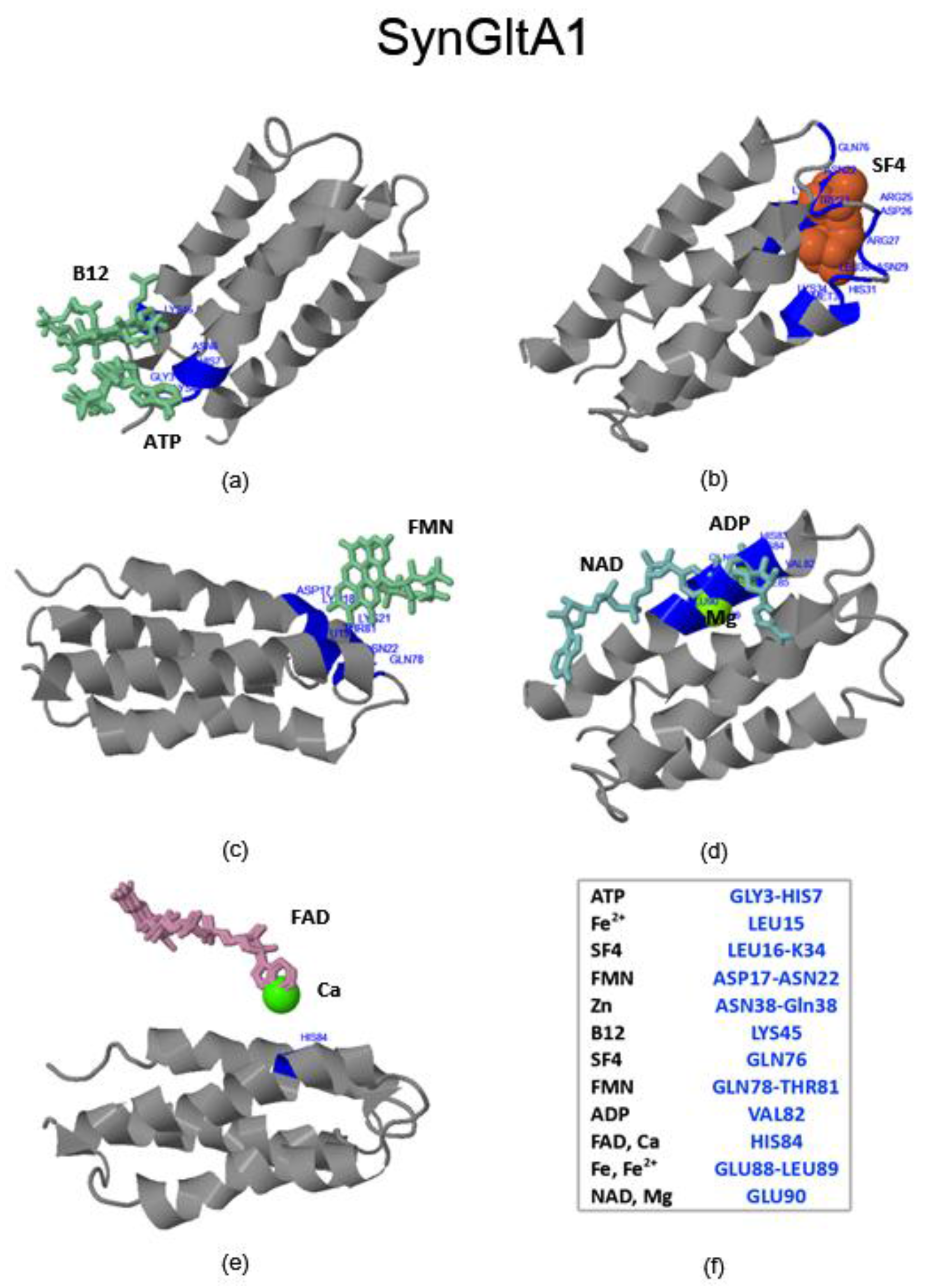
2.3. Virtual Spectroscopy and 3D Ligand Binding Prediction of Hecht_α (SynRescue) Proteins
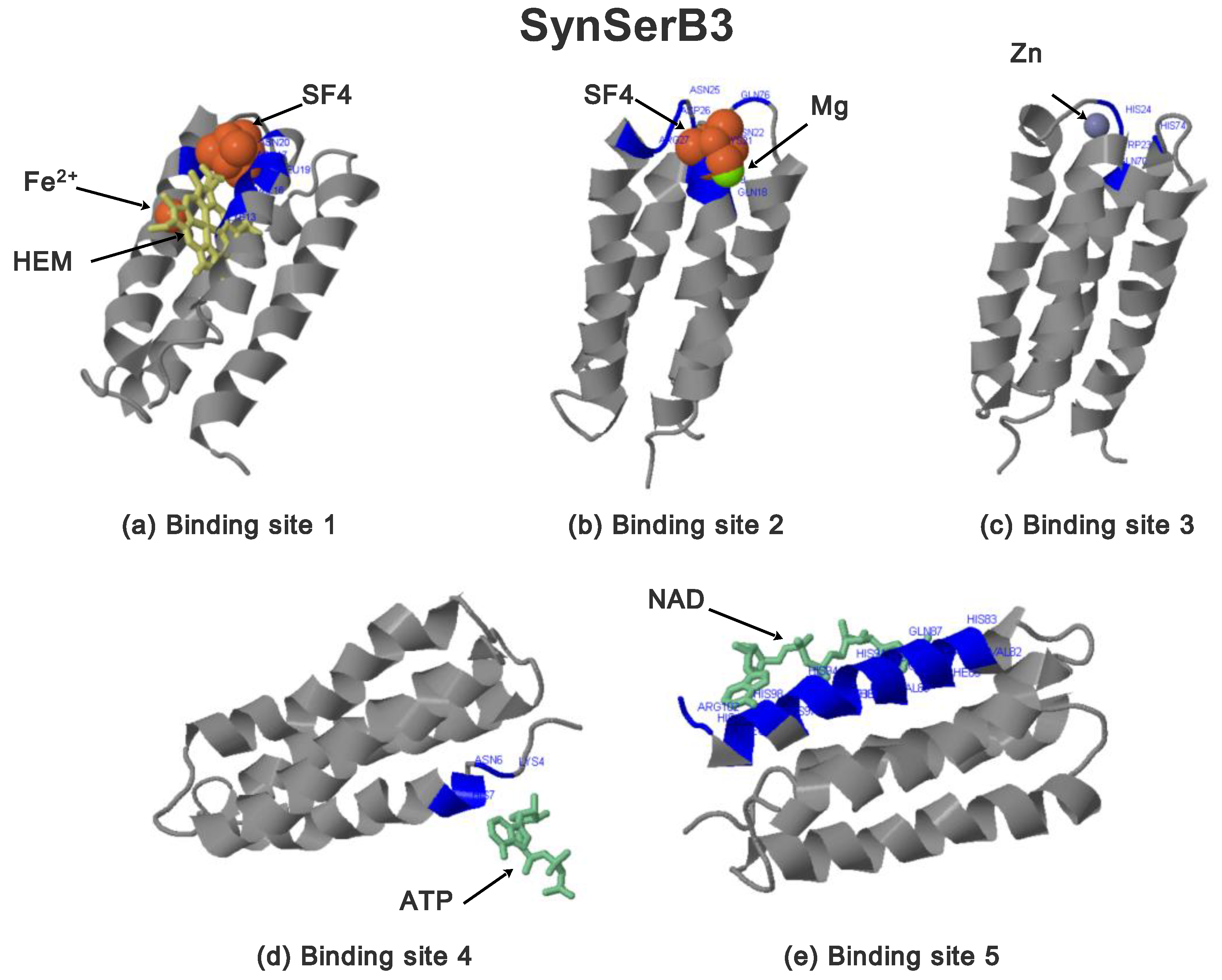
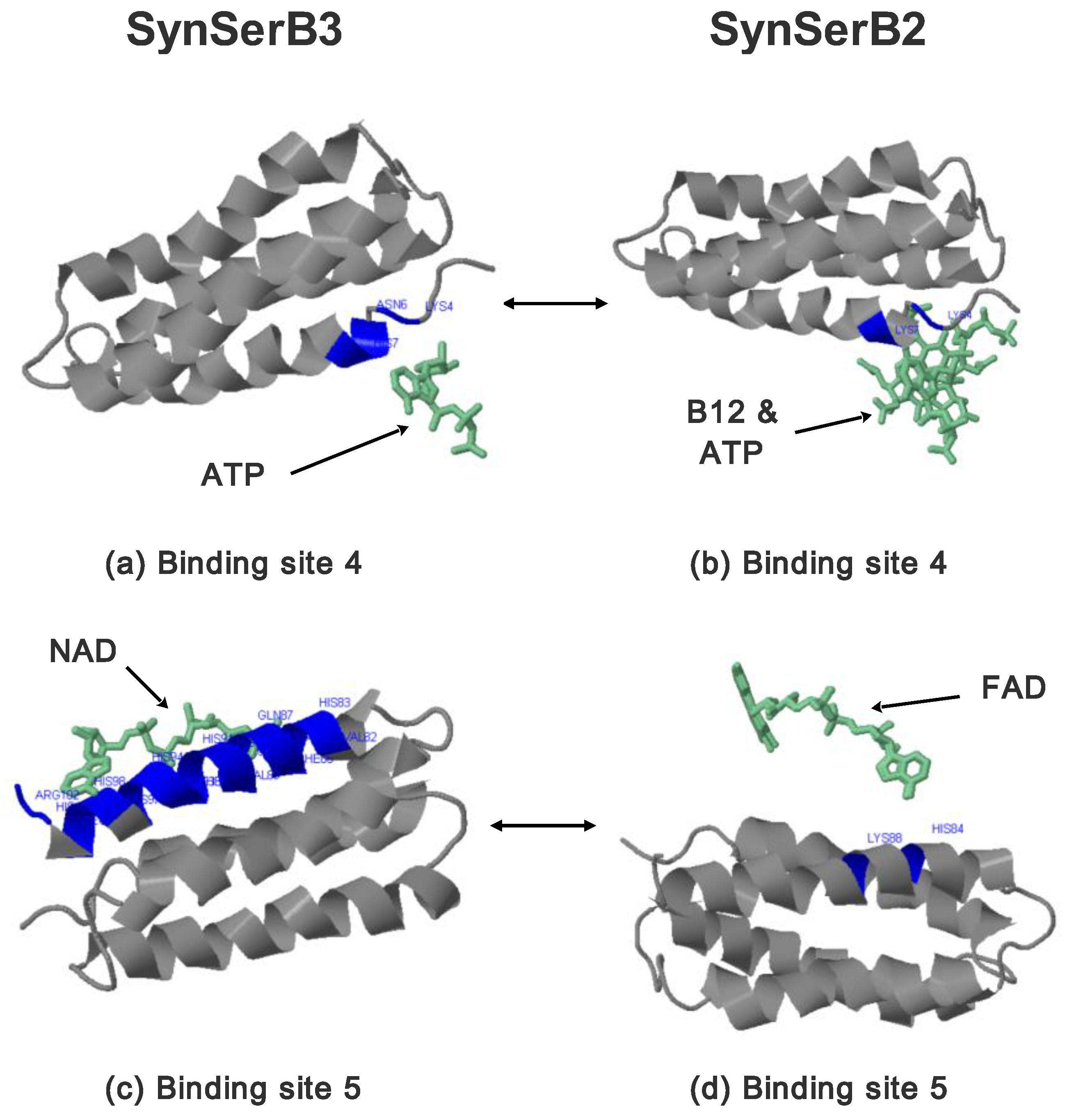
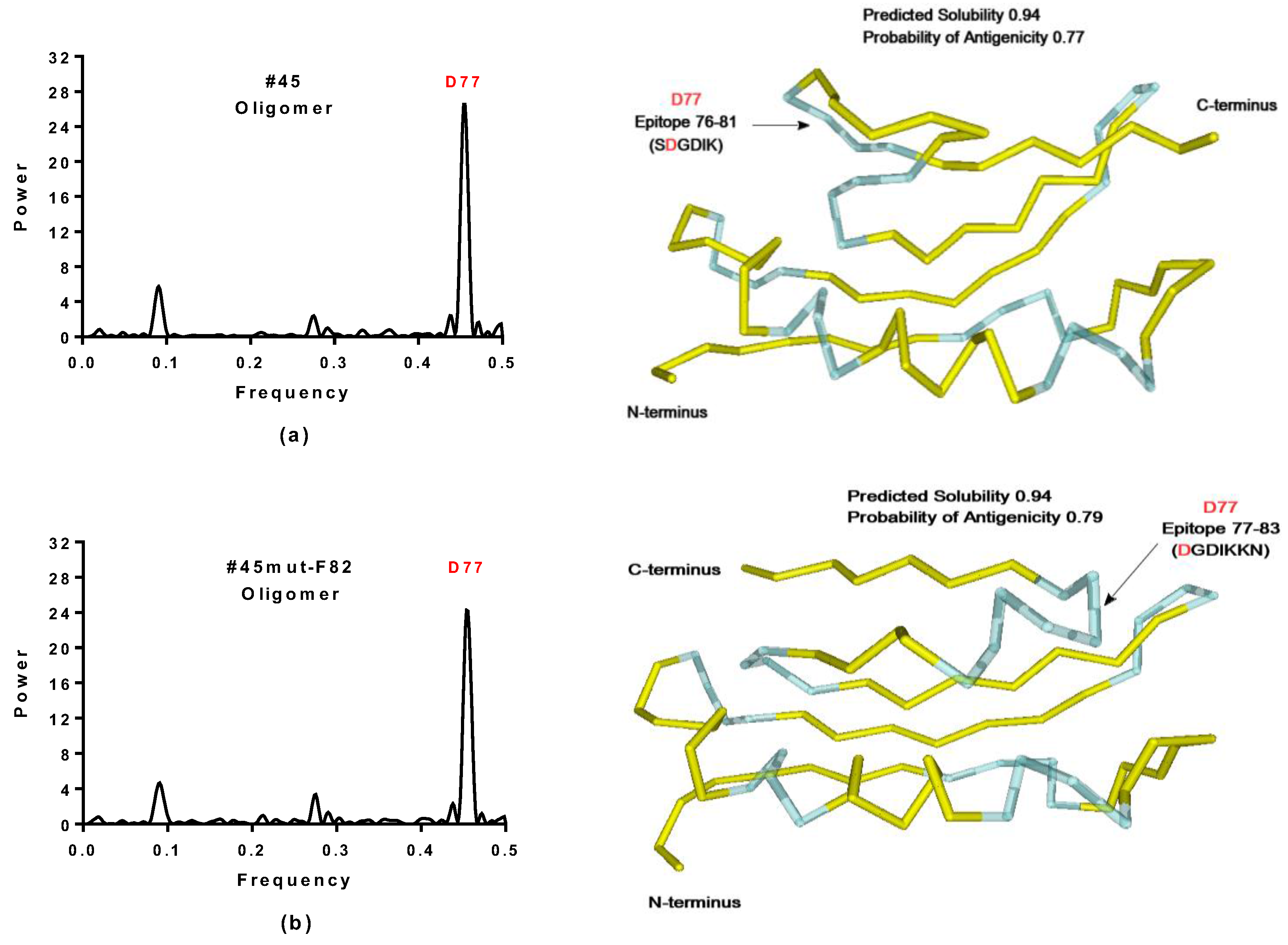
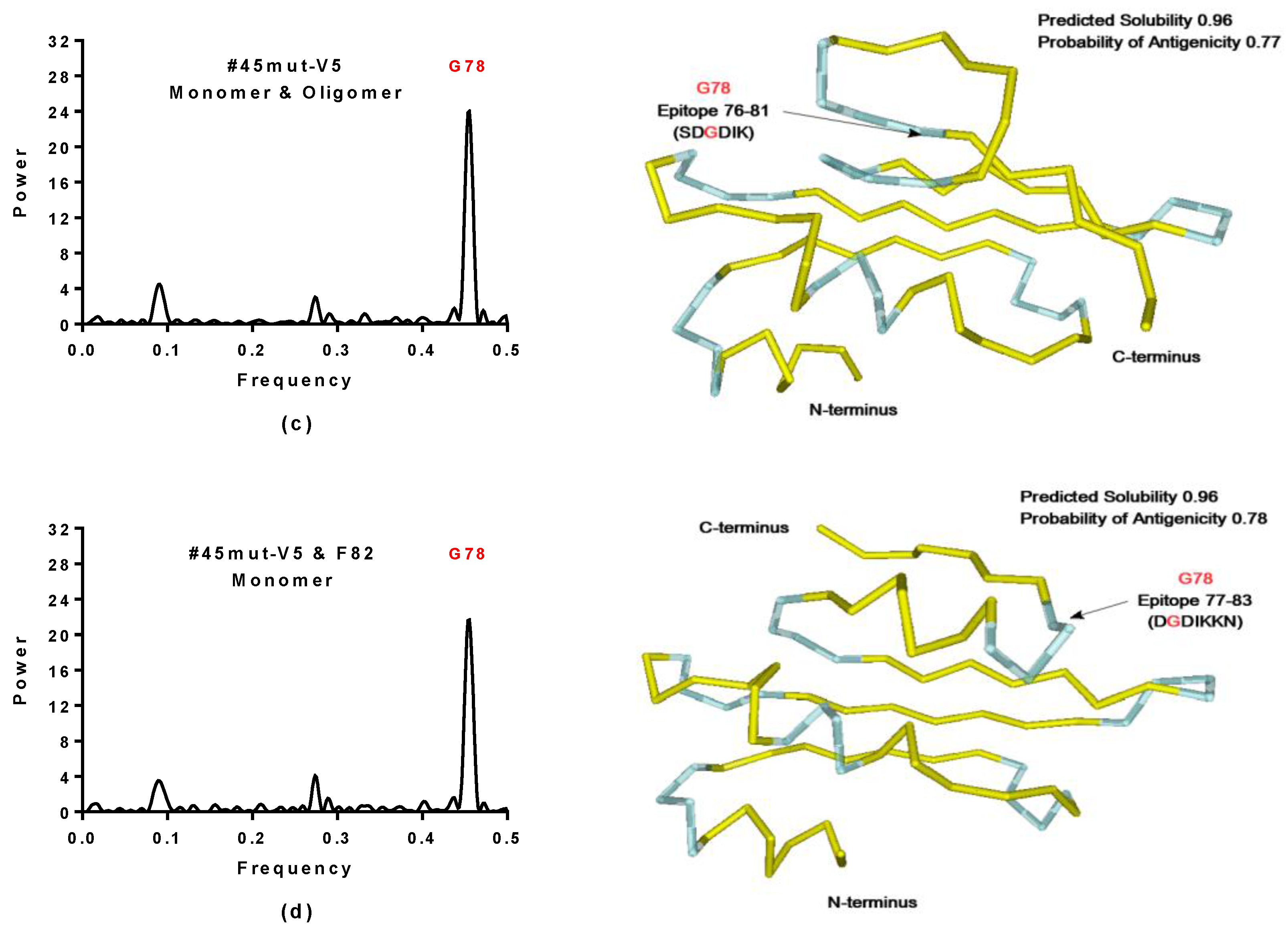
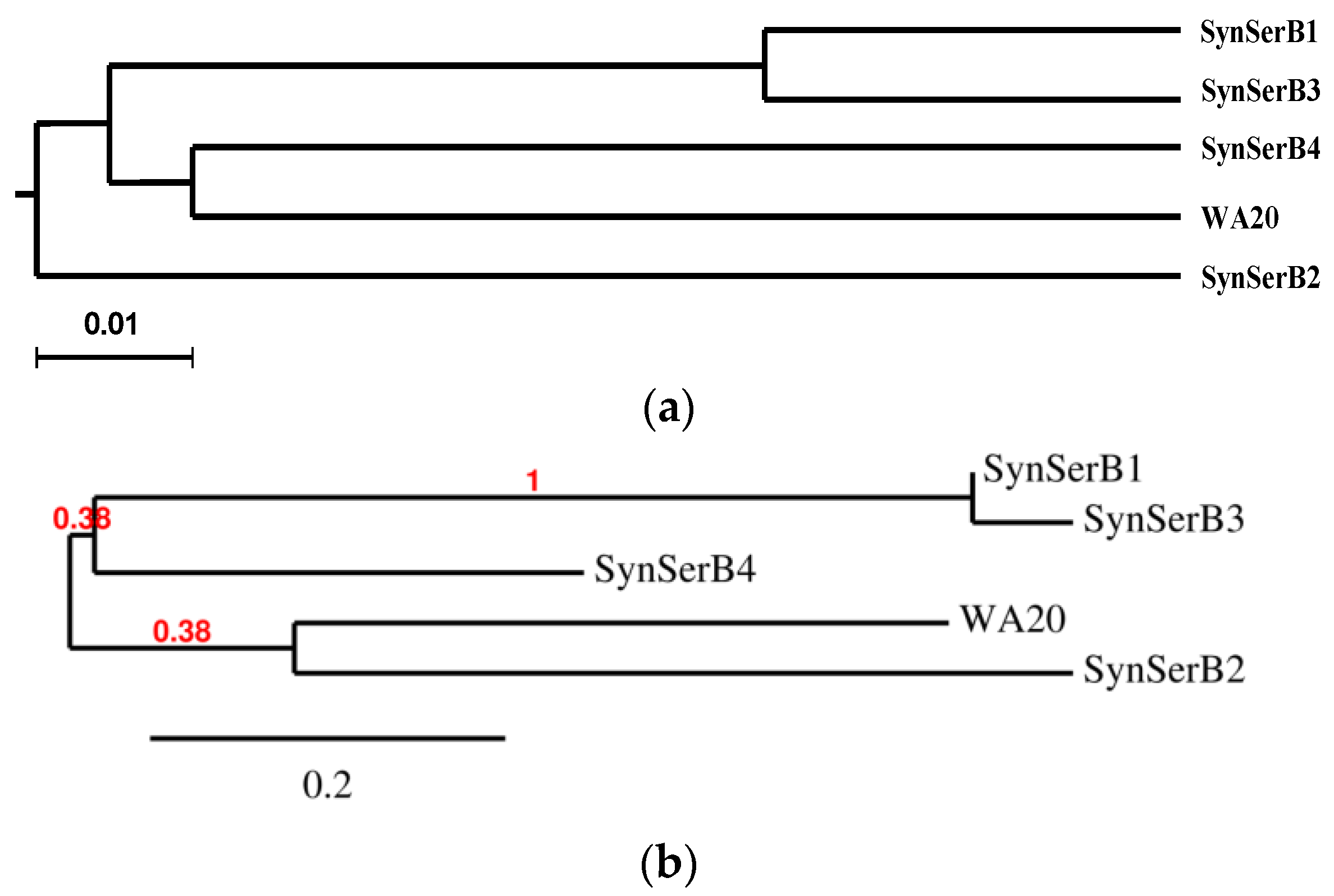
2.3.1. SynSerB Rescue Proteins
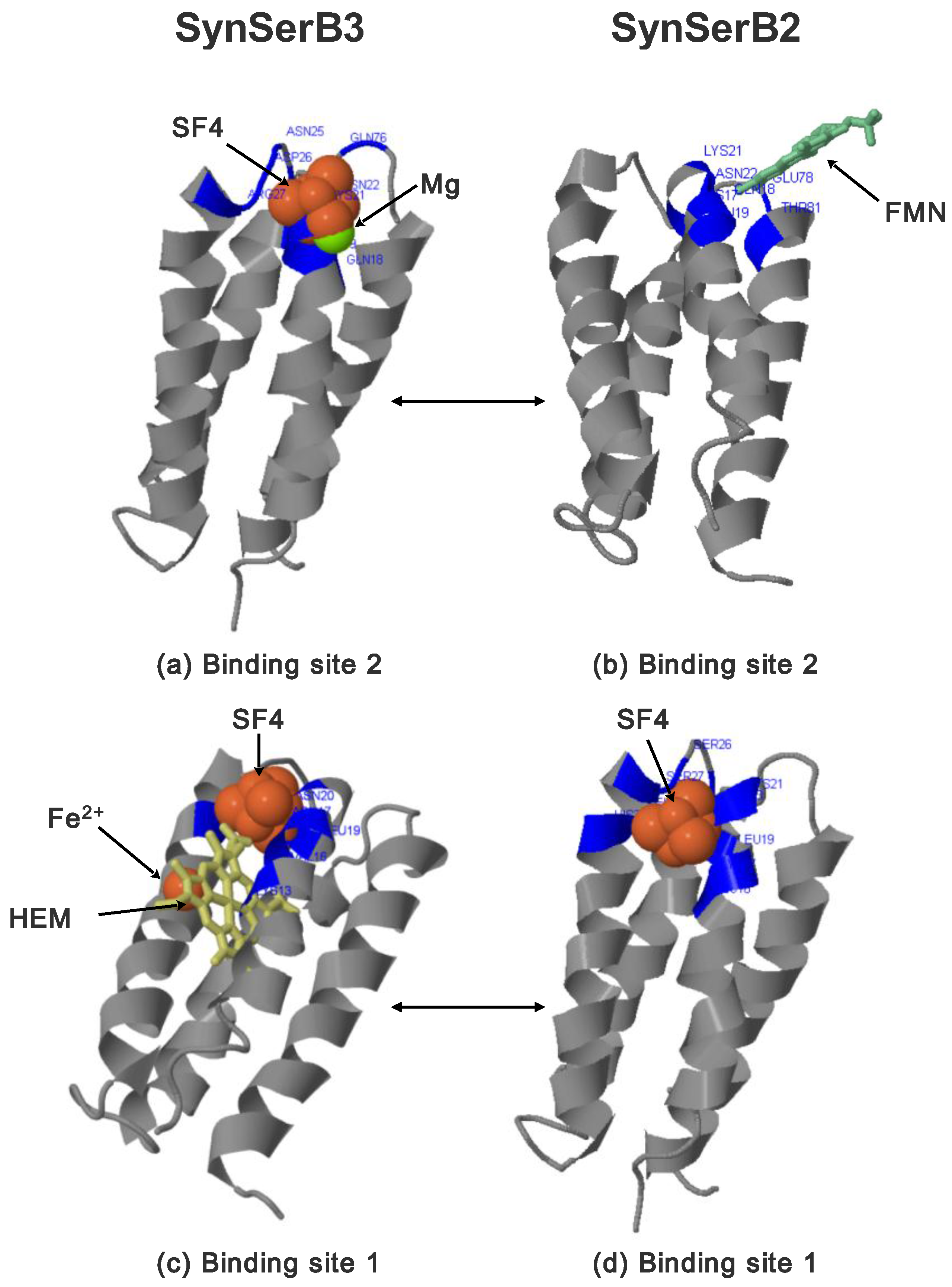
- FMN interaction with binding site 2 shifts the binding of SF4 to binding site 1, but the additional interaction with HEM (heme) and Fe2+ is missing (region 16–31, Figure 8c,d);
- the heterogen B12 in the SynSerB2 mutant disrupts the binding of ATP at binding site 4 (amino acid positions 4–7, Figure 9a,b);
- the heterogen FAD in SynSerB2 mutant disrupts the binding of NAD at binding site 5 (amino acid positions 82–102, Figure 9c,d).
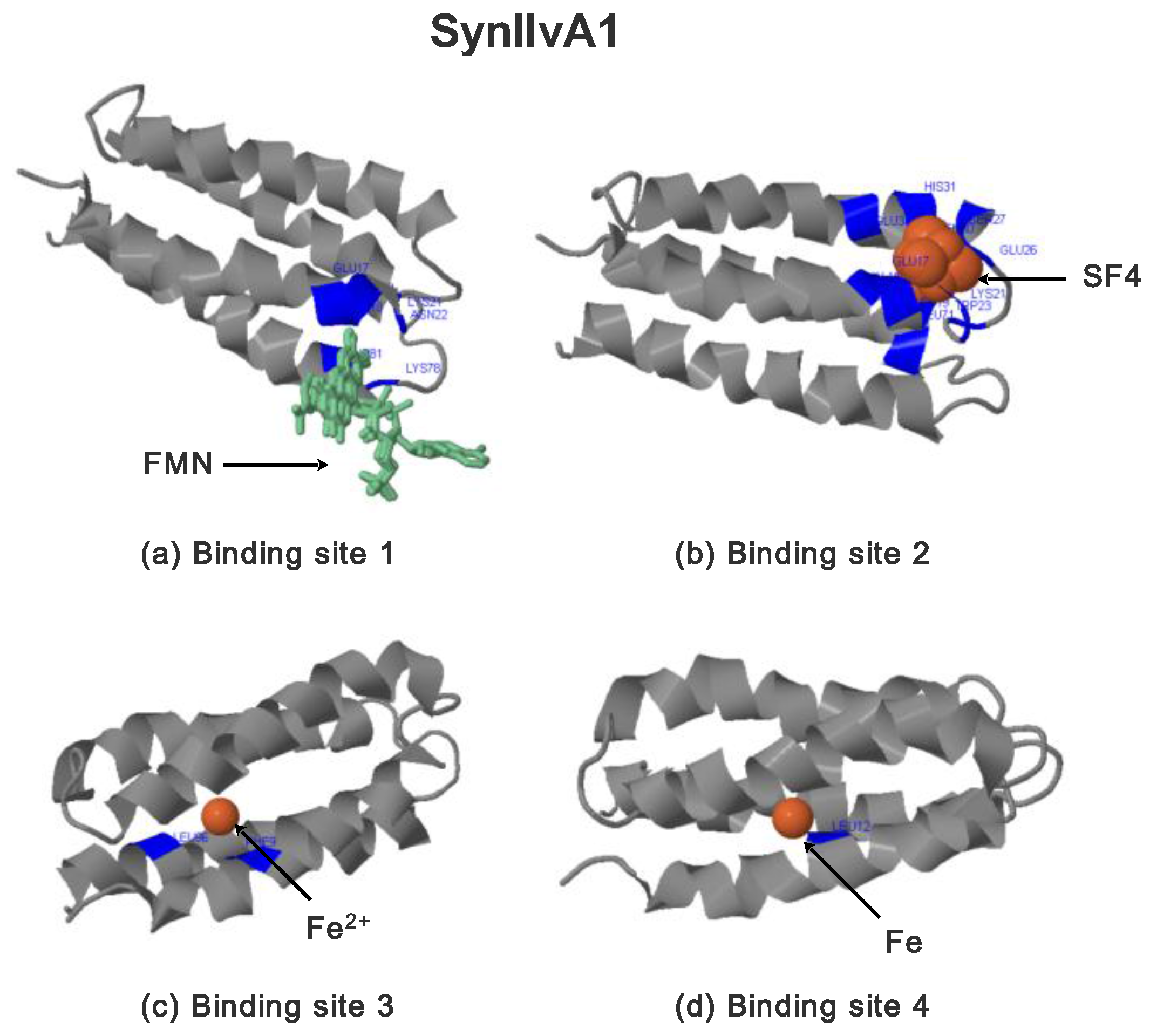
2.3.2. SynIlvA Rescue Proteins
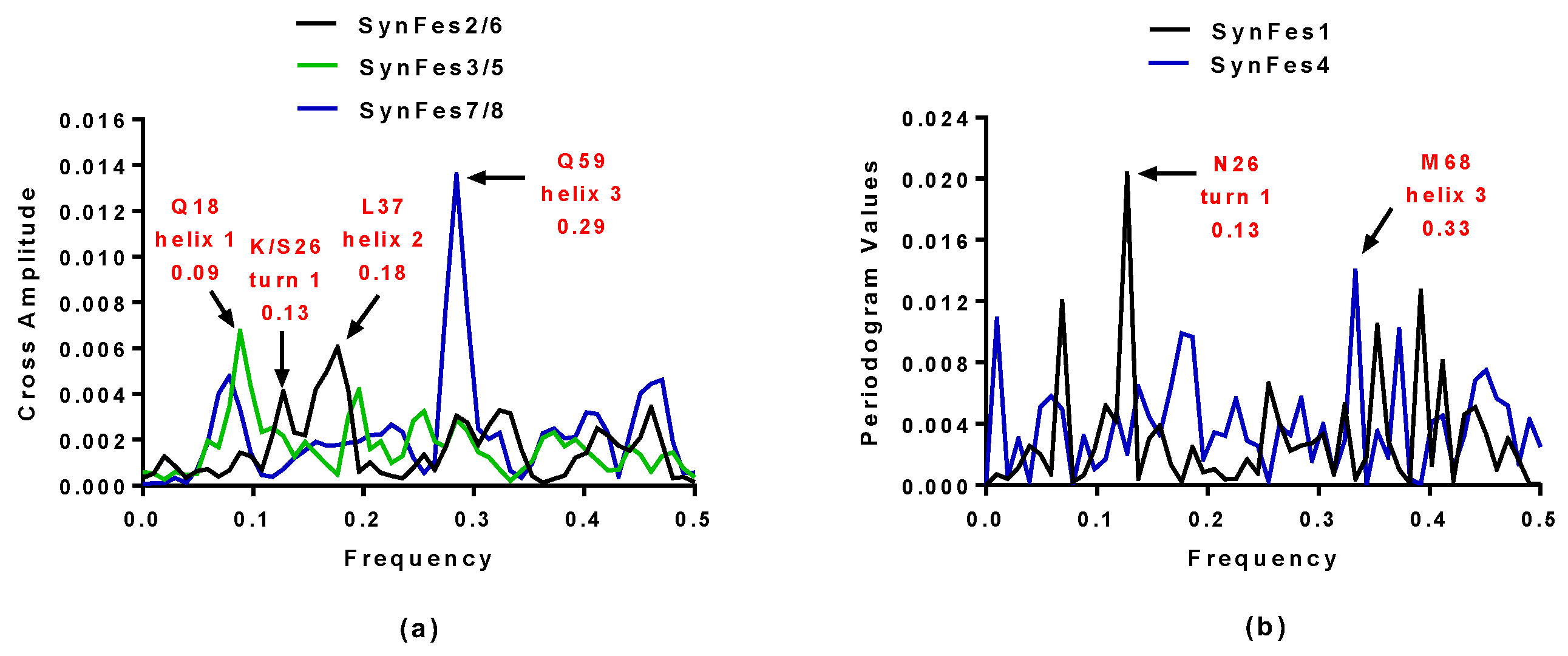
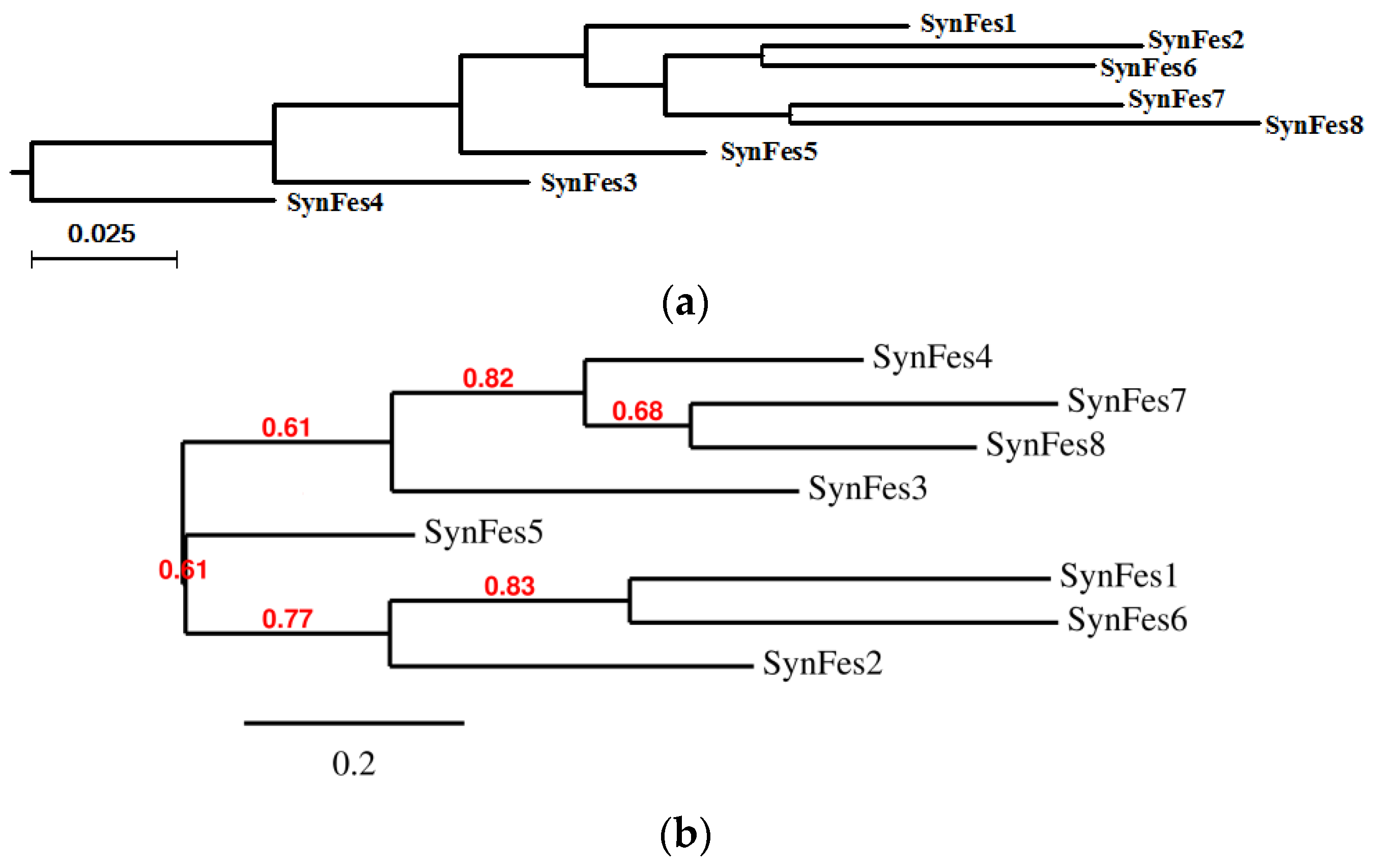
2.3.3. SynFes and SynGltA Rescue Proteins
- in addition to the positions 13 (Fe/Fe2+) and 49 (Fe2+) shared by SynFes6 and SynFes2, SynFes2 has two extra Fe heterogen binding positions at amino acid sites 64 and 96;
- at position Q49 in SynFes6, FAD and B12 could disrupt the binding of other heterogens (Fe2+, ATP, NAD, GAL, MAN and GLC), which is not the case for SynFes2;
- SynFes2 has two additional binding sites for heterogen Ca at positions 1 and 49.
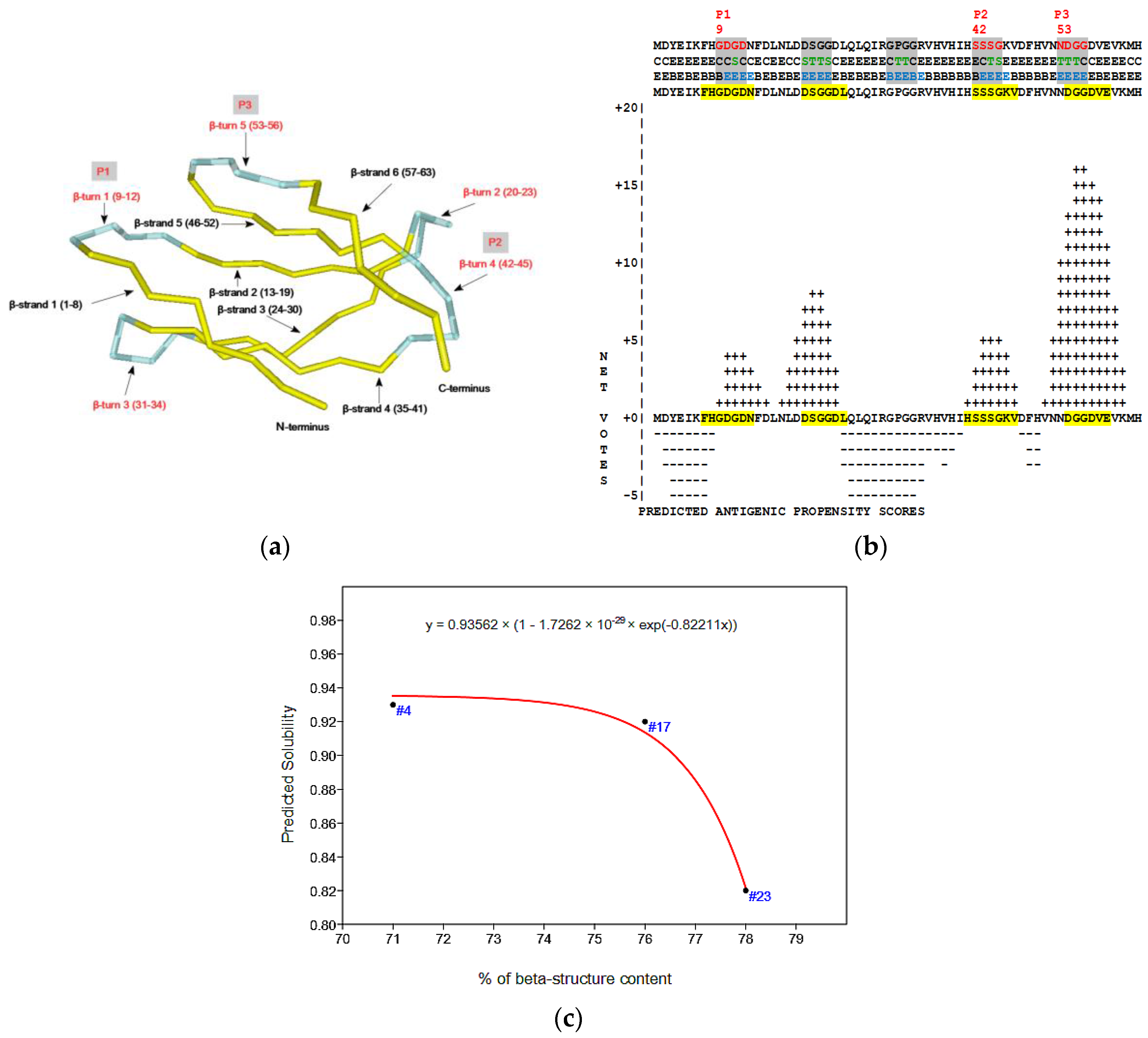
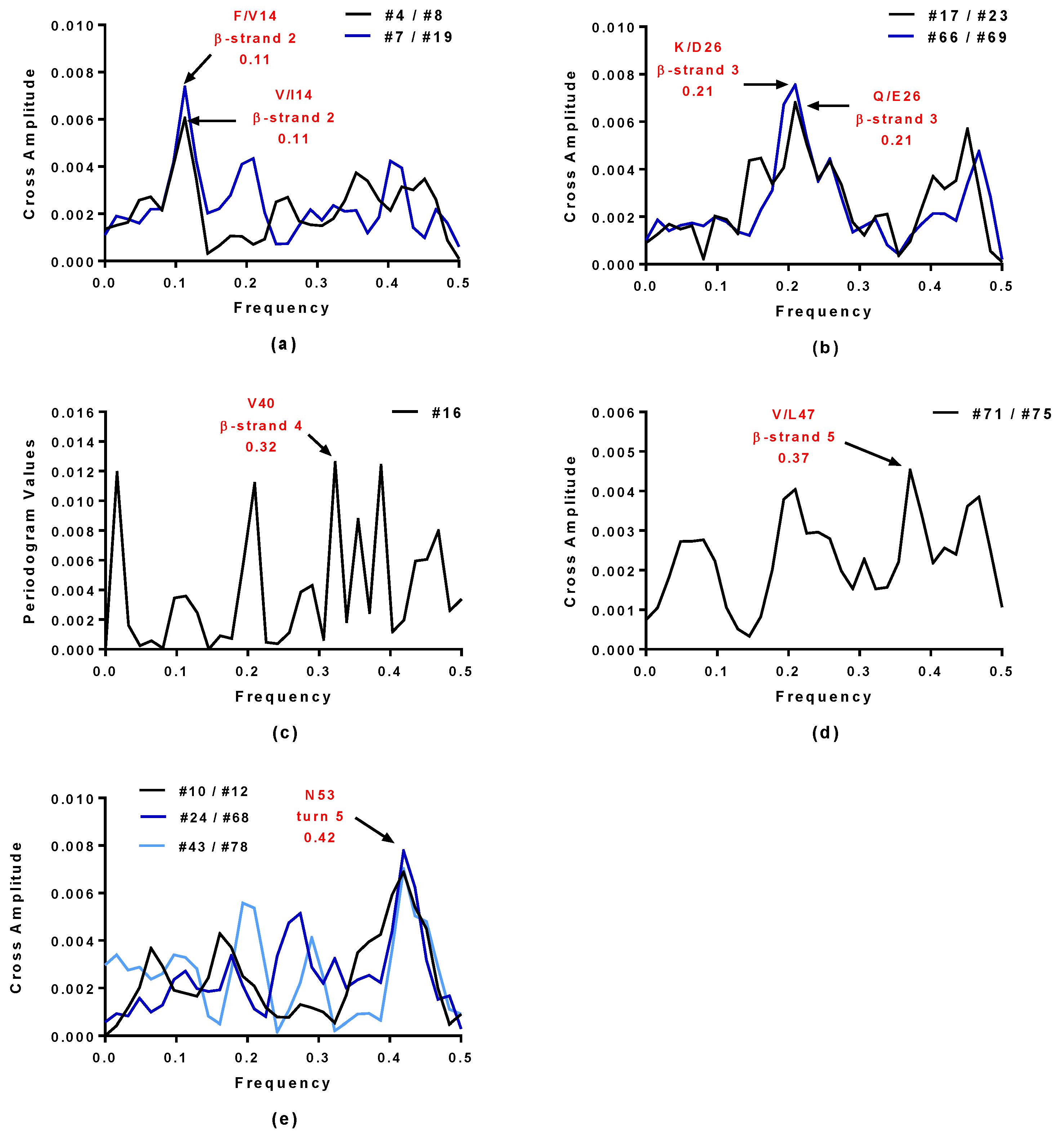
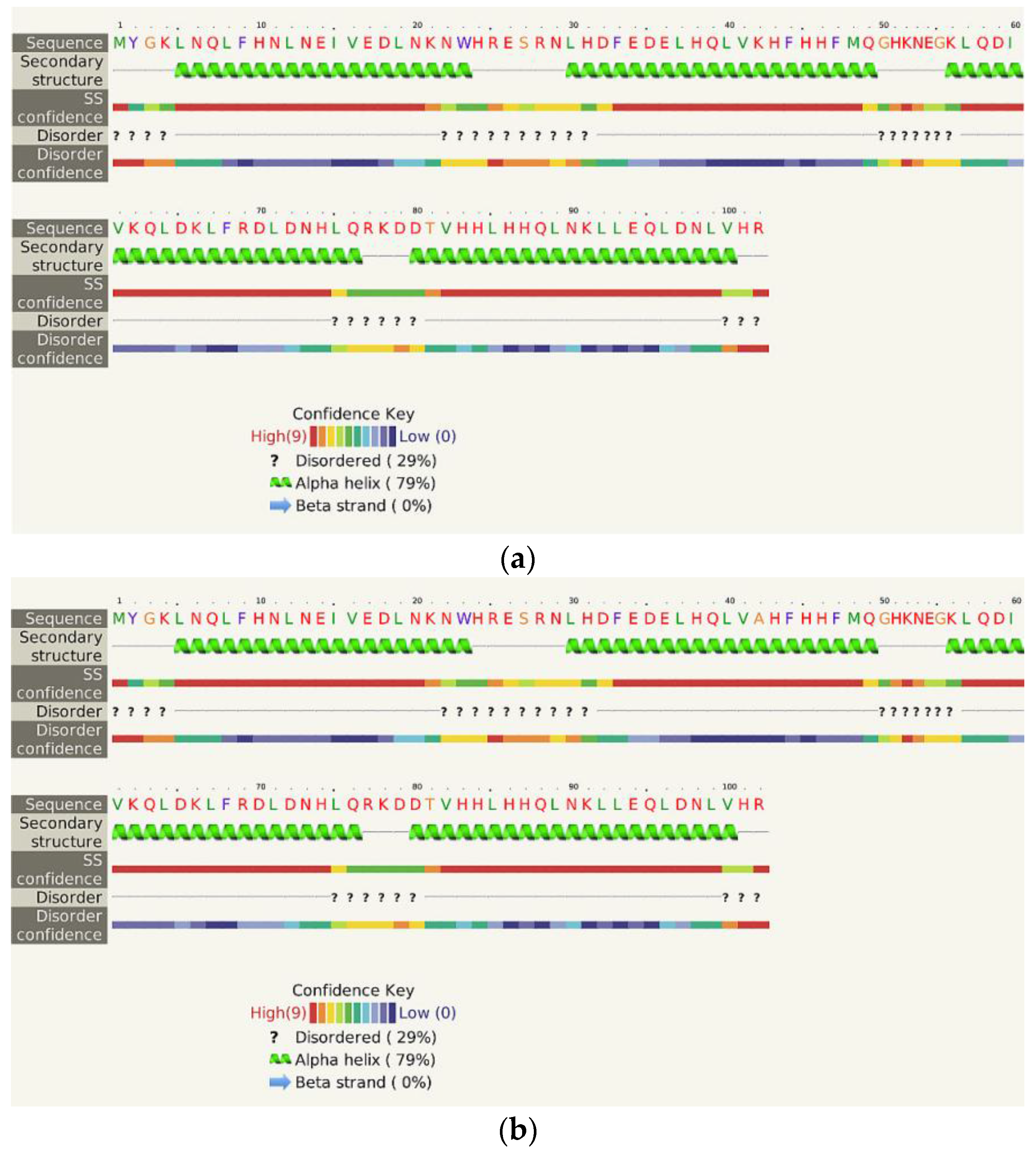
2.4. Virtual Spectroscopy and 3D Structure Prediction of Hecht_β Proteins
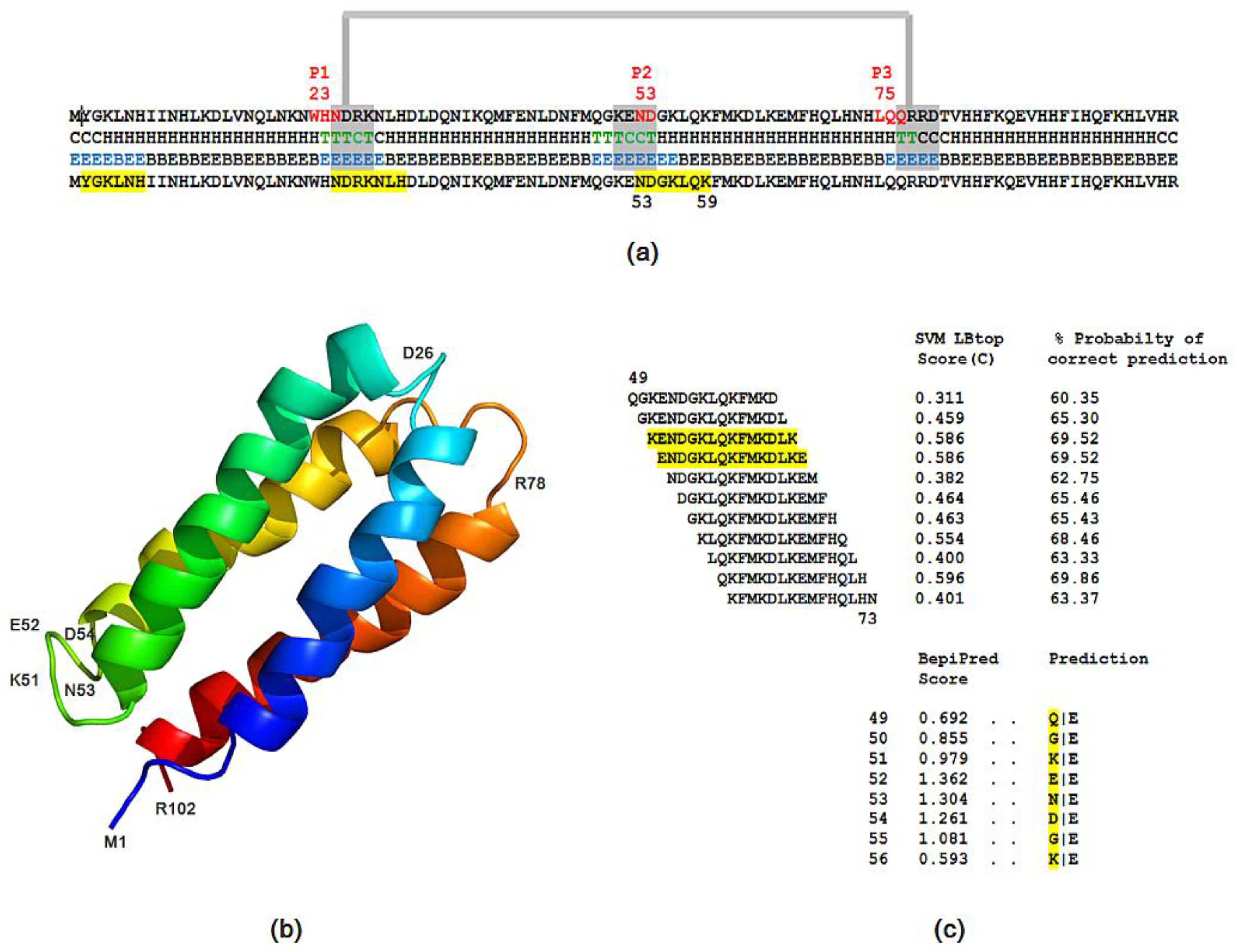
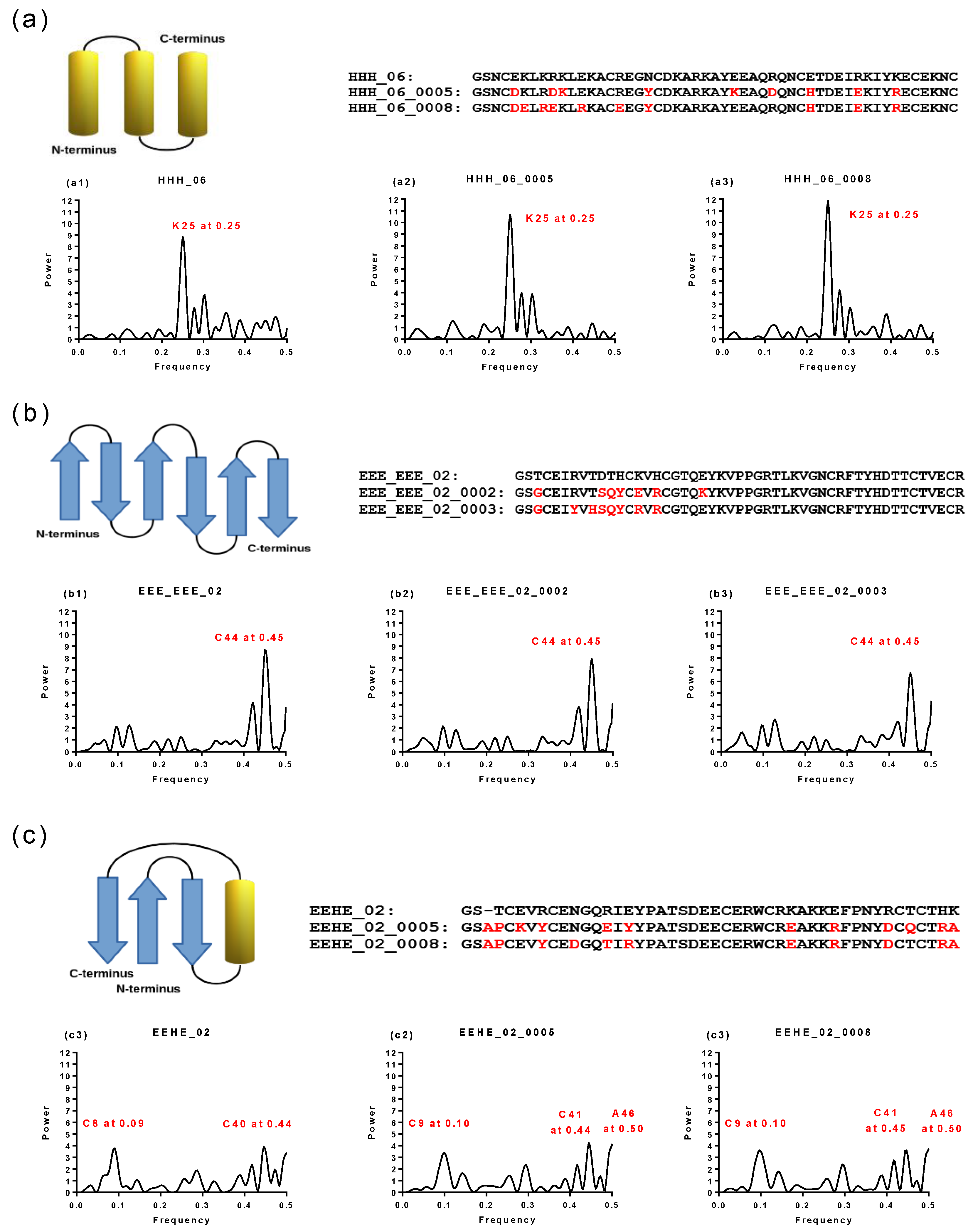
2.5. Structural and Functional Characterization of Hecht_α and Hecht_β Proteins
3. Materials and Methods
3.1. Protein Datasets
- VAN (V = A, C, G) was used to encode six polar residues (H, Q, N, K, D, E) and
- NTN (N = A, T, C, G) was used to encode five nonpolar residues (F, L, I, M, V).
3.2. Spectral Analysis
3.3. Bioinformatic Software Tools Used for Sequence Analyzes
3.3.1. Hydrophobicity Profiles
3.3.2. Solubility, Antigenicity, Surface Accessibility, 2D/3D and Tree Structure Predictions
- The probability of protein antigenicity was determined by ANTIGENpro, a sequence-based, alignment-free and pathogen-independent predictor of the protein antigenicity (Table A3) [31]. Prediction of linear B-cell epitopes was carried out using COBEpro, BepiPred and LBotope servers (Figure 3c) [32,33,34].
- The Phyre2 server could not predict the 3D structure of the Hecht_β proteins because the models were insufficiently valid. The confidence was considered too low (<70%) for submission to 3DLigandSite [37]. The FOLDpro method was used for protein fold recognition and template-based 3D structure prediction (Figure 4a, Figure A4) of all β-proteins [52]. The protein 2D and 3D structures were presented using Unipro UGENE software [71]. PDB files of the #17 and #45 models are supplied as Schemes S33–S37.
4. Conclusions
- improvement of the existing tools for protein structure and function analysis,
- new algorithms for the construction of de novo protein subsets and
- additional information on the complex natural sequence space and its relation to the individual subspaces of de novo sequences.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First (5') Letter | Second Letter | Third (3') Letter | |||
|---|---|---|---|---|---|
| U/T | C | A | G | ||
| U/T | Phe (F) | Ser (S) | Tyr (Y) | Cys (C) | U/T |
| Phe (F) | Ser (S) | Tyr (Y) | Cys (C) | C | |
| Leu (L) | Ser (S) | stop | stop | A | |
| Leu (L) | Ser (S) | stop | Trp (W) | G | |
| C | Leu (L) | Pro (P) | His (H) | Arg (R) | U/T |
| Leu (L) | Pro (P) | His (H) | Arg (R) | C | |
| Leu (L) | Pro (P) | Gln (Q) | Arg (R) | A | |
| Leu (L) | Pro (P) | Gln (Q) | Arg (R) | G | |
| A | Ile (I) | Thr (T) | Asn (N) | Ser (S) | U/T |
| Ile (I) | Thr (T) | Asn (N) | Ser (S) | C | |
| Ile (I) | Thr (T) | Lys (K) | Arg (R) | A | |
| Met (M) | Thr (T) | Lys (K) | Arg (R) | G | |
| G | Val (V) | Ala (A) | Asp (D) | Gly (G) | U/T |
| Val (V) | Ala (A) | Asp (D) | Gly (G) | C | |
| Val (V) | Ala (A) | Glu (E) | Gly (G) | A | |
| Val (V) | Ala (A) | Glu (E) | Gly (G) | G | |
| Amino Acid | Abbreviation | Cornette Scale 1 | Kyte–Doolittle Scale | EIIP (Ry) |
|---|---|---|---|---|
| Phenylalanine | Phe (F) | 0.140 | 2.8 | 0.0946 |
| Leucine | Leu (L) | 0.000 | 3.8 | 0.0000 |
| Valine | Val (V) | 0.114 | 4.2 | 0.0057 |
| Isoleucine | Ile (I) | 0.102 | 4.5 | 0.0000 |
| Methionine | Met (M) | 0.164 | 1.9 | 0.0823 |
| Serine | Ser (S) | 0.699 | −0.8 | 0.0829 |
| Proline | Pro (P) | 0.903 | −1.6 | 0.0198 |
| Alanine | Ala (A) | 0.622 | 1.8 | 0.0373 |
| Threonine | Thr (T) | 0.865 | −0.7 | 0.0941 |
| Cysteine | Cys (C) | 0.182 | 2.5 | 0.0829 |
| Tryptophan | Trp (W) | 0.528 | −0.9 | 0.0548 |
| Arginine | Arg (R) | 0.485 | −4.5 | 0.0959 |
| Glycine | Gly (G) | 0.648 | −0.4 | 0.0050 |
| Tyrosine | Tyr (Y) | 0.278 | −1.3 | 0.0516 |
| Histidine | His (H) | 0.595 | −3.2 | 0.0242 |
| Glutamine | Gln (Q) | 0.970 | −3.5 | 0.0761 |
| Glutamic acid | Glu (E) | 0.854 | −3.5 | 0.0058 |
| Asparagine | Asn (N) | 0.701 | −3.5 | 0.0036 |
| Aspartic acid | Asp (D) | 1.000 | −3.5 | 0.1263 |
| Lysine | Lys (K) | 0.995 | −3.9 | 0.0371 |
| Synthetic Proteins | Predicted Antigenicity 1 | Predicted Solubility 2 | Predicted Solubility 3 |
|---|---|---|---|
| Hecht_α | |||
| SynSerB1 | 0.65 | Soluble (0.94) | Medium (15.28) |
| SynSerB2 | 0.65 | Soluble (0.78) | Medium (15.34) |
| SynSerB3 | 0.56 | Soluble (0.93) | Medium (14.75) |
| SynSerB4 | 0.45 | Soluble (0.97) | Medium (15.30) |
| SynGltA1 | 0.52 | Soluble (0.92) | Medium (15.17) |
| SynIlvA1 | 0.75 | Soluble (0.51) | Medium (16.86) |
| SynIlvA2 | 0.78 | Insoluble (0.51) | Medium (16.38) |
| SynFes1 | 0.83 | Soluble (0.90) | Medium (15.42) |
| SynFes2 | 0.57 | Insoluble (0.58) | Medium (15.37) |
| SynFes3 | 0.63 | Soluble (0.62) | Medium (15.91) |
| SynFes4 | 0.81 | Soluble (0.50) | Medium (16.85) |
| SynFes5 | 0.55 | Soluble (0.75) | Medium (15.29) |
| SynFes6 | 0.57 | Soluble (0.92) | Medium (15.14) |
| SynFes7 | 0.42 | Soluble (0.98) | Medium (15.40) |
| SynFes8 | 0.84 | Soluble (0.69) | Medium (16.70) |
| Hecht_β | |||
| #4 | 0.84 | Soluble (0.93) | Medium (17.76) |
| #7 | 0.71 | Soluble (0.90) | Medium (20.04) |
| #8 | 0.66 | Soluble (0.82) | Medium (18.71) |
| #10 | 0.58 | Soluble (0.90) | Medium (16.49) |
| #12 | 0.66 | Soluble (0.94) | Medium (16.91) |
| #16 | 0.71 | Soluble (0.96) | Medium (16.32) |
| #17 | 0.79 | Soluble (0.92) | Medium (18.24) |
| #19 | 0.80 | Soluble (0.84) | Medium (17.33) |
| #23 | 0.74 | Soluble (0.82) | Medium (19.57) |
| #24 | 0.66 | Soluble (0.91) | Medium (16.49) |
| #43 | 0.65 | Soluble (0.88) | Medium (20.50) |
| #66 | 0.59 | Soluble (0.93) | Medium (18.71) |
| #68 | 0.32 | Soluble (0.94) | Medium (19.18) |
| #69 | 0.47 | Soluble (0.87) | Medium (18.27) |
| #71 | 0.38 | Soluble (0.78) | Medium (15.72) |
| #75 | 0.51 | Soluble (0.80) | Medium (17.58) |
| #78 | 0.75 | Soluble (0.92) | Medium (21.33) |
| Hecht_β Protein | Predicted Epitopes N-Terminus 1 | Predicted Epitopes Central 1 | Predicted Epitopes C-Terminus 1 |
|---|---|---|---|
| #4 | 2 | 0 | 1 ‡ |
| #7 | 2 | 0 | 2 |
| #8 | 1 | 0 | 2 |
| #10 | 2 | 0 | 1 † |
| #12 | 1 | 0 | 1 † |
| #16 | 2 | 0 | 1 † |
| #17 | 2 | 0 | 2 |
| #19 | 2 | 0 | 1 † |
| #23 | 2 | 0 | 2 |
| #24 | 2 | 0 | 1 † |
| #43 | 2 | 0 | 2 |
| #66 | 2 | 0 | 2 |
| #68 | 2 | 0 | 2 |
| #69 | 1 | 0 | 2 |
| #71 | 2 | 0 | 2 |
| #75 | 2 | 0 | 2 |
| #78 | 2 | 0 | 2 |








References
- Huang, P.-S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, D.N.; Bartlett, G.J.; Burton, A.J.; Heal, J.W.; Niitsu, A.; Thomson, A.R.; Wood, C.W. De novo protein design: How do we expand into the universe of possible protein structures? Curr. Opin. Struct. Biol. 2015, 33, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.S.; Greisman, J.B.; Hecht, M.H. De novo proteins with life-sustaining functions are structurally dynamic. J. Mol. Biol. 2016, 428, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, D.N.; Bartlett, G.J.; Bruning, M.; Thomson, A.R. New currency for old rope: From coiled-coil assemblies to α-helical barrels. Curr. Opin. Struct. Biol. 2012, 22, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Boyle, A.I.; Bruning, M.; Bartlett, G.J.; Vincent, T.L.; Zaccai, N.R.; Armstrong, C.T.; Bromley, E.H.C.; Booth, P.J.; Brady, R.L.; et al. A basis set of de novo coiled-coil peptide oligomers for rational protein design and synthetic biology. ACS Synth. Biol. 2012, 1, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.A.; McKinley, K.L.; Bradley, L.H.; Viola, S.R.; Hecht, M.H. De novo designed proteins from a library of artificial sequences function in Escherichia coli and enable cell growth. PLoS ONE 2011, 6, e15364. [Google Scholar] [CrossRef] [PubMed]
- Ó Conchúir, S.; Barlow, K.A.; Pache, R.A.; Ollikainen, N.; Kundert, K.; O’Meara, M.J.; Smith, C.A.; Kortemme, T. A web resource for standardized benchmark datasets, metrics, and Rosetta protocols for macromolecular modeling and design. PLoS ONE 2015, 10, e0130433. [Google Scholar] [CrossRef] [PubMed]
- West, M.W.; Hecht, M.H. Binary patterning of polar and nonpolar amino acids in the sequences and structures of native proteins. Protein Sci. 1995, 4, 2032–2039. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.D.; Das, A.; Go, A.; Bradley, L.H.; Wei, Y. De novo proteins from designed combinatorial libraries. Protein Sci. 2004, 13, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Hecht, H. Novel proteins: From fold to function. Curr. Opin. Chem. Biol. 2011, 15, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Digianantonio, K.M.; Hecht, M.H. A protein constructed de novo enables cell growth by altering gene regulation. Proc. Natl. Acad. Sci. USA 2016, 113, 2400–2405. [Google Scholar] [CrossRef] [PubMed]
- West, M.W.; Wang, W.; Patterson, J.; Mancias, J.D.; Beasley, J.R.; Hecht, M.H. De novo amyloid proteins from designed combinatorial libraries. Proc. Natl. Acad. Sci. USA 1999, 96, 11211–11216. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, N.; Konjevoda, P.; Gotovac, N. A new rule-based system for the construction and structural characterization of artificial proteins. In Chaos and Complex Systems; Stavrinides, S., Banerjee, S., Caglar, H., Ozer, M., Eds.; Springer: Berlin, Germany, 2013; pp. 95–103. [Google Scholar]
- Good, I.G. Analyzing the Large Number of Variables in Biomedical and Satellite Imagery, 1st ed.; Wiley: Hoboken, NJ, USA, 2011; pp. 1–3. [Google Scholar]
- Bradley, L.H.; Thumfort, P.P.; Hecht, M.H. De novo proteins from binary-patterned combinatorial libraries. In Protein Design: Methods and Applications, 1st ed.; Guerois, R., López de la Paz, M., Eds.; Humana Press: Totowa, NJ, USA, 2007; Volume 340, pp. 53–69. [Google Scholar]
- Wei, Y.; Liu, T.; Sazinsky, S.L.; Moffet, D.A.; Pelczer, I.; Hecht, M.H. Stably folded de novo proteins from a designed combinatorial library. Protein Sci. 2003, 12, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Moffet, D.A.; Hecht, M.H. De novo proteins from combinatorial libraries. Chem. Rev. 2001, 101, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Kamtekar, S.; Schiffer, J.M.; Xiong, H.; Babik, J.M.; Hecht, M.H. Protein design by binary patterning of polar and non-polar amino acids. Science 1993, 262, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Roy, S.; Hecht, M.H. Screening combinatorial libraries of de novo proteins by hydrogen-deuterium exchange and electrospray mass spectrometry. J. Am. Chem. Soc. 1999, 121, 9509–9513. [Google Scholar] [CrossRef]
- Wang, W.; Hecht, M.H. Rationally designed mutations convert de novo amyloid-like fibrils into monomeric beta-sheet proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 2760–2765. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Yanase, K.; Sato, T.; Unzai, S.; Hecht, M.H.; Arai, R. Self-assembling nano-architectures created from a protein nano-building block using an intermolecularly folded dimeric de novo protein. J. Am. Chem. Soc. 2015, 137, 11285–11293. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, N.; Konjevoda, P. Prediction of secondary protein structure with binary coding patterns of amino acid and nucleotide physicochemical properties. Int. J. Quant. Chem. 2003, 92, 123–134. [Google Scholar] [CrossRef]
- Thomas, F.; Burgess, N.C.; Thomson, A.R.; Woolfson, D.N. Controlling the assembly of coiled–coil peptide nanotubes. Angew. Chem. Int. Ed. 2016, 55, 987–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, N.C.; Sharp, T.H.; Thomas, F.; Wood, C.W.; Thomson, A.R.; Zaccai, N.R.; Brady, L.; Serpell, L.C.; Woolfson, D.N. Modular design of self-assembling peptide-based nanotubes. J. Am. Chem. Soc. 2015, 137, 10554–10562. [Google Scholar] [CrossRef] [PubMed]
- Mahendran, K.R.; Niitsu, A.; Kong, L.; Thomson, A.R.; Sessions, R.B.; Woolfson, D.N.; Bayley, H. A monodisperse transmembrane α-helical peptide barrel. Nat. Chem. 2016. [Google Scholar] [CrossRef]
- Bhardwaj, G.; Mulligan, V.K.; Bahl, D.C.; Gilmore, J.M.; Harvey, P.J.; Cheneval, O.; Buchko, G.; Pulavarti, S.; Kaas, Q.; Eletsky, A.; et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016, 538, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Cornette, J.L.; Cease, K.B.; Margalit, H.; Spouge, J.L.; Berzofsky, J.A.; DeLisi, C. Hydrophobicity scales and computational techniques for detecting amphipathic structures in proteins. J. Mol. Biol. 1987, 195, 659–685. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Magnan, C.N.; Baldi, P. SSpro/ACCpro 5: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnan, C.N.; Zeller, M.C.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Sweredoski, M.J.; Pierre Baldi, P. COBEpro: A novel system for predicting continuous B-cell epitopes. Protein Eng. Des. Sel. 2009, 22, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Lund, O.; Morten Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.; Ansari, H.R.; Raghava, P.S.G. Improved method for linear B-cell epitope prediction using antigen’s primary sequence. PLoS ONE 2013, 8, e62216. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Li, C.; Webb, G.I.; Tey, B.; Song, J.; Ramanan, R.N. Periscope: quantitative prediction of soluble protein expression in the periplasm of Escherichia coli. Sci. Rep. 2016, 6, 21844. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J. 3DLigandSite: Predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010, 38, W469–W473. [Google Scholar] [CrossRef] [PubMed]
- Wass, M.N.; Sternberg, M.J. Prediction of ligand binding sites using homologous structures and conservation at CASP8. Proteins 2009, 77 (Suppl. S9), 147–151. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, N.; Glisic, S.; Prljic, J.; Perovic, V.; Botta, M.; Veljkovic, V. Discovery of new therapeutic targets by the informational spectrum method. Curr. Protein Pept. Sci. 2008, 9, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Manetti, F.; Veljkovic, N.; Perovic, V.; Vercammen, J.; Hayes, S.; Massa, S.; Witvrouw, M.; Debyser, Z.; Veljkovic, V.; et al. Novel virtual screening protocol based on the combined use of molecular modeling and electron-ion interaction potential techniques to design HIV-1 integrase inhibitors. J. Chem. Inf. Model. 2007, 47, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Cosic, I. The Resonant Recognition Model of Macromolecular Bioactivity: Theory and Applications; Birkhäuser: Basel, Switzerland, 1997; pp. 1–87. [Google Scholar]
- Veljkovic, V.; Veljkovic, N.; Esté, J.A.; Hüther, A.; Dietrich, U. Application of the EIIP/ISM bioinformatics concept in development of new drugs. Curr. Med. Chem. 2007, 14, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, N.; Manojlović, Z.; Turčić, P.; Martinić, R.; Konjevoda, P.; Weitner, T.; Wardega, P.; Gabričević, M. A simple three-step method for design and affinity testing of new antisense peptides: An example of Erythropoietin. Int. J. Mol. Sci. 2014, 15, 9209–9223. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Cornette, J.L.; Margalit, H.; Berzofsky, J.A.; DeLisi, C. Periodic variation in side-chain polarities of T-cell antigenic peptides correlates with their structure and activity. Proc. Natl. Acad. Sci. USA 1995, 92, 8368–8372. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.P.; Tweedy, N.B.; Williams, R.W.; Richardson, J.S.; Richardson, D.C. Betadoublet: De novo design, synthesis, and characterization of a 3-sandwich protein. Proc. Natl. Acad. Sci. USA 1994, 91, 8747–8751. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Pochan, D.J.; Ozbas, B.; Rajagopal, K.; Pakstis, L.; Kretsinger, J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; van der Schaft, D.V.J.; Barendsz-Janson, A.F.; Cox, A.; Boudier, H.A.J.S.; Hillen, H.F.P.; Mayo, K.H. Anginex, a designed peptide that inhibits angiogenesis biochem. Biochem. J. 2001, 354, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Brown, F.; Doughan, G.; Hoey, E.M.; Martin, S.J.; Rima, B.K.; Trudgett, A. Vaccine Design; Wiley: Chichester, UK, 1993; pp. 33–44. [Google Scholar]
- Cheng, J.; Baldi, P. A machine learning information retrieval approach to protein fold recognition. Bioinformatics 2006, 22, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Ratanji, K.D.; Derrick, J.P.; Rebecca, J.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.C.; Bradley, L.H.; Jinadasa, S.P.; Hecht, M.H. Cofactor binding and enzymatic activity in an unevolved superfamily of de novo designed 4-helix bundle proteins. Protein Sci. 2009, 18, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.C.; Hecht, M.H. Directed evolution of the peroxidase activity of a de novo-designed protein. Protein Eng. Des. Sel. 2012, 25, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shi, Y. Metabolic enzymes and coenzymes in transcription—A direct link between metabolism and transcription? Trends Genet. 2004, 20, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Hassan, I. Design, principles, network architecture and their analysis strategies as applied to biological systems. In Systems Biology Application in Synthetic Biology; Singh, S., Ed.; Springer: New Delhi, India, 2016; pp. 21–31. [Google Scholar]
- Perovic, V.R.; Muller, C.P.; Niman, H.L.; Veljkovic, N.; Dietrich, U.; Tosic, D.D.; Glisic, S.; Veljkovic, V. Novel phylogenetic algorithm to monitor human tropism in Egyptian H5N1-HPAIV reveals evolution toward efficient human-to-human transmission. PLoS ONE 2013, 8, e61572. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereeper, A.; Audic, S.; Claverie, J.-M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evolut. Biol. 2010, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Cicchillo, R.M.; Baker, M.A.; Schnitzer, E.J.; Newman, E.B.; Krebs, C.; Booker, S.J. Escherichia coli L-serine deaminase requires a [4Fe-4S] cluster in catalysis. J. Biol. Chem. 2004, 279, 32418–32425. [Google Scholar] [CrossRef] [PubMed]
- Digianantonio, K.M.; Korolev, M.; Hecht, M.H. A non-natural protein rescues cells deleted for a key enzyme in central metabolism. ACS Synth. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Young, D. Computational Drug Design, 1st ed.; Wiley: Hoboken, NJ, USA, 2009; pp. 1–5. [Google Scholar]
- Higgs, P.G.; Attwood, T.K. Bioinformatics and Molecular Evolution, 1st ed.; Blackwell: Malden, MA, USA, 2005; pp. 1–10. [Google Scholar]
- Petoukhov, S.V. The system-resonance approach in modeling genetic structures. Biosystems 2016, 139, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Montano, M.A.; Coronel-Brizio, H.F.; Hernández-Montoya, A.R.; Ramos-Fernández, A. Codon information value and codon transition-probability distributions in short-term evolution. Physica A 2016, 454, 117–128. [Google Scholar] [CrossRef]
- StatSoft, Inc. STATISTICA (Data Analysis Software System), Version 7. 2014. Available online: http://www.statsoft.com (accessed on 31 July 2016).
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Hocke, K.; Kämpfer, N. Gap filling and noise reduction of unevenly sampled data by means of the Lomb-Scargle periodogram. Atmos. Chem. Phys. 2009, 9, 4197–4206. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Agyepong, K.; Serpedin, E.; Dougherty, E.R. Detecting periodic genes from irregularly sampled gene expressions: A comparison study. EURASIP J. Bioinform. Syst. Biol. 2008, 769293. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, N.; Konjevoda, P.; Manojlović, Z.; Štambuk, A.; Turčić, P.; Gotovac, N. Synthetic proteins designed using ternary coding patterns: From nucleotide information to protein structure, function and music. Symmetry Cult. Sci. 2016, 27, 163–171. [Google Scholar]















| Synthetic Proteins | Frequency Peak 1 | Amino Acid/No. |
|---|---|---|
| Hecht_α proteins (SynRescue) | ||
| SynSer (B1 to B4) | 0.28 | Q58 |
| SynGltA1 | 0.28 | Q58 |
| SynIlvA (1 and 2) | 0.28 | Q58 |
| SynFes (1 to 8) | 0.28 | Q58 |
| Hecht_β-proteins | ||
| #4, #7, #23, #43 | 0.45 | E57 |
| #66, #68, #69, #75 | 0.45 | E57 |
| #8, #10, #12, #16 | 0.45 | D57 |
| #17, #19, #24, #71, #78 | 0.45 | D57 |
| Synthetic Protein | Spectral Analysis 1 (Fourier Single Series) | Spectral Analysis 1 (LSSA) | Amino Acid/No. | Activity 2 (Cell Growth) |
|---|---|---|---|---|
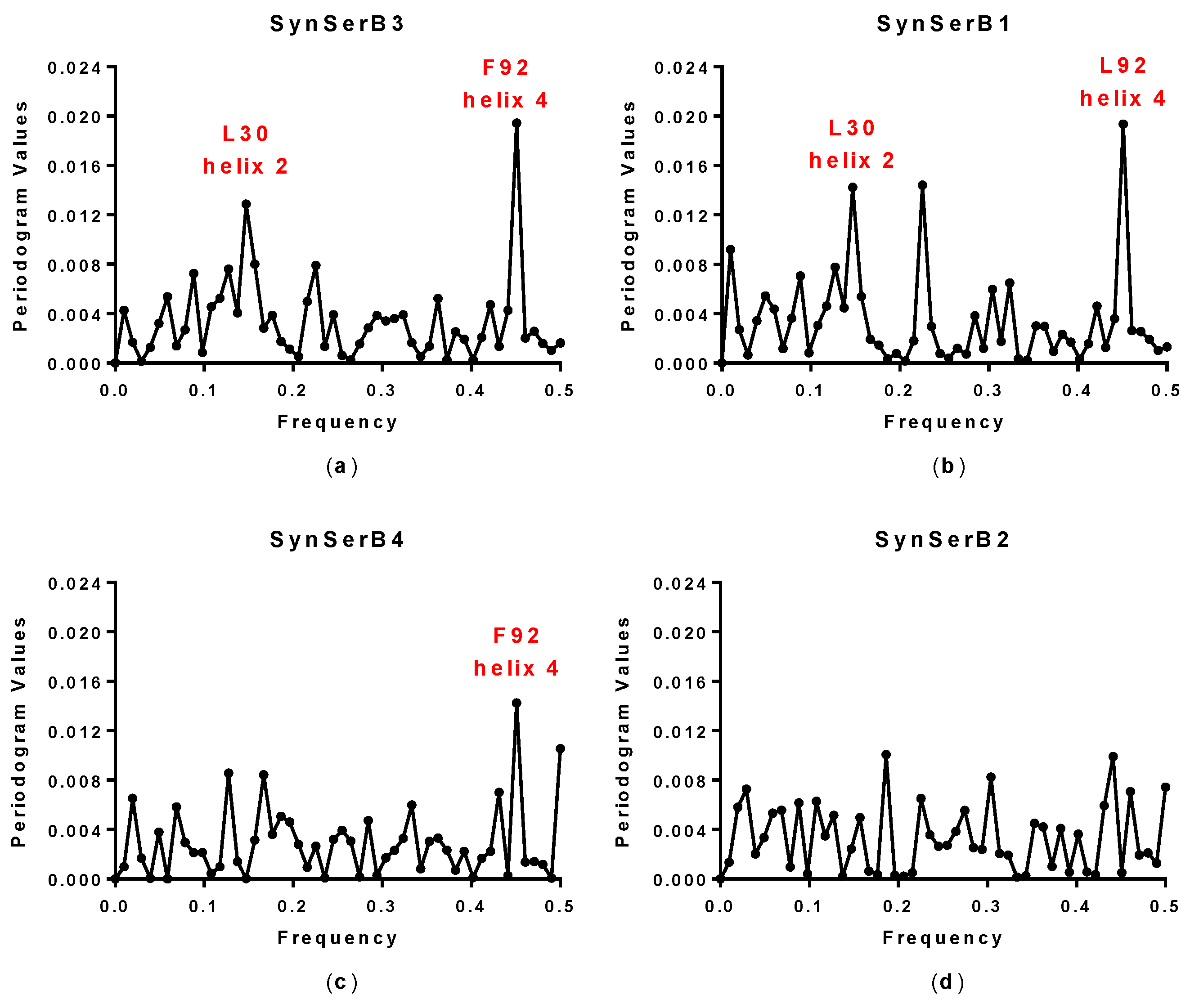
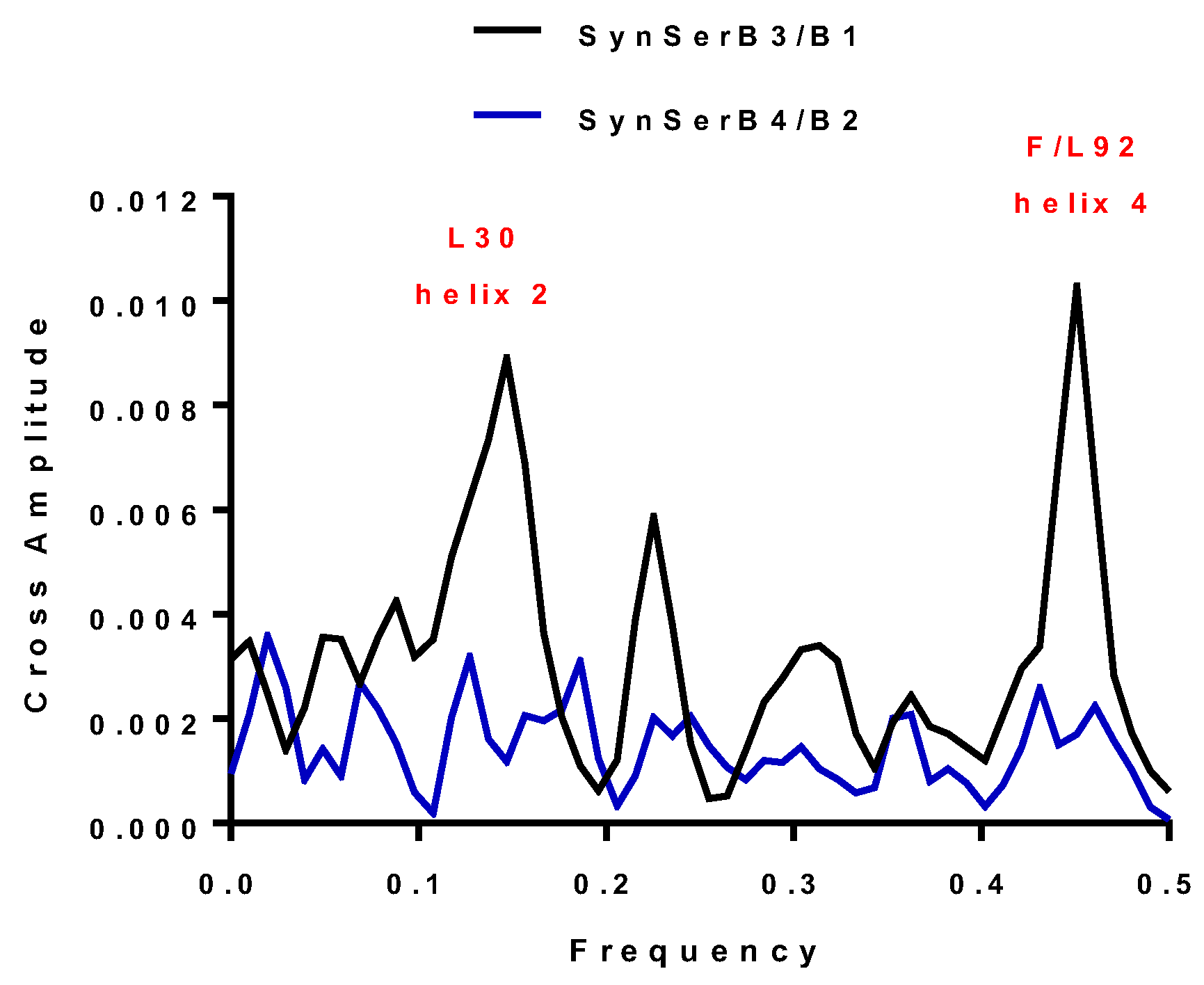
| SynSerB3 | 0.15 and 0.45 | 0.15 and 0.45 | L30 and F92 | ++ |
| SynSerB1 | 0.15 and 0.45 | 0.15 and 0.45 | L30 and L92 | ++ |
| SynSerB4 | 0.45 | 0.45 | M92 | + |
| SynSerB2 | - | - | - | + |
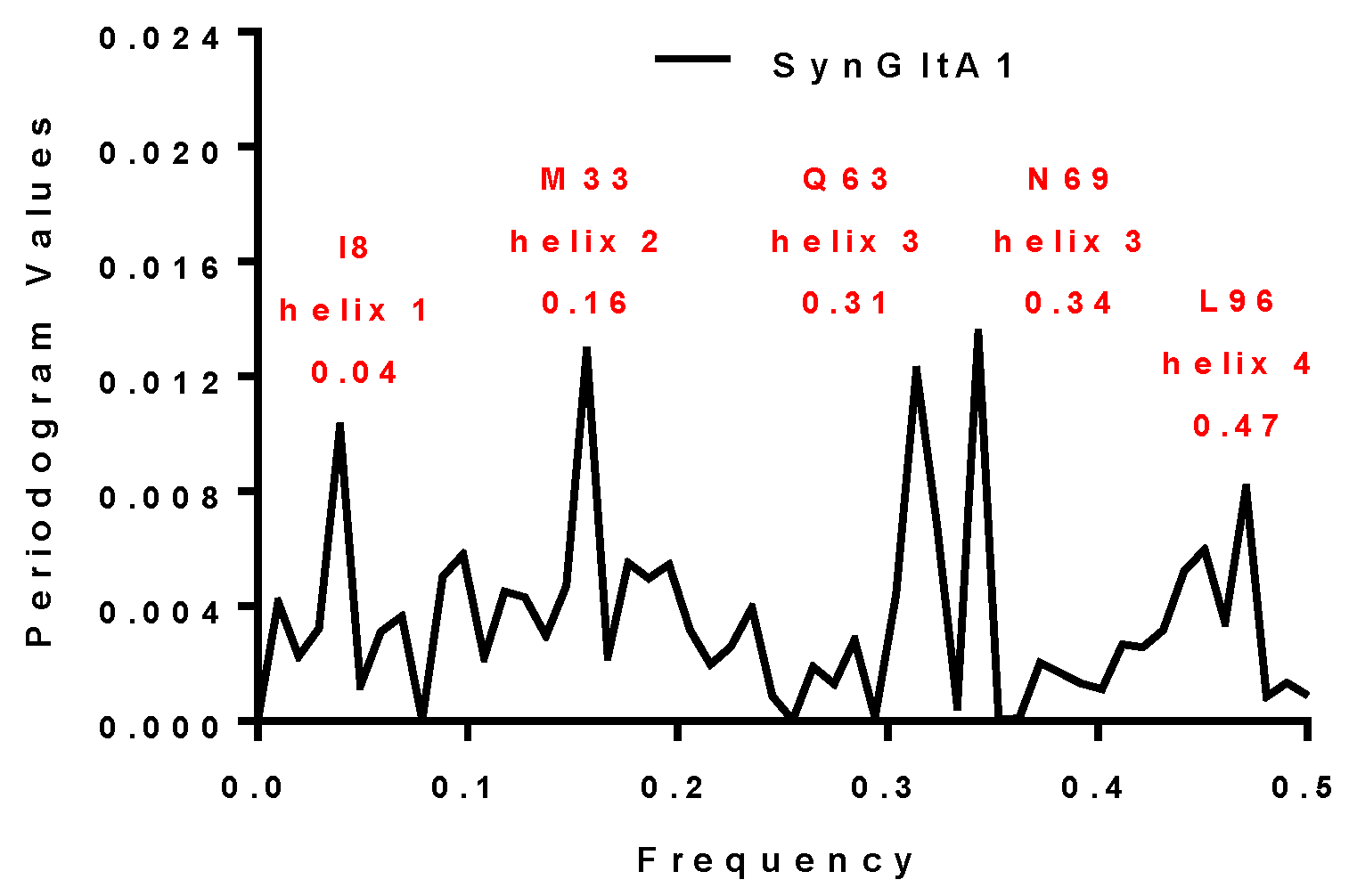
| SynGltA1 | 0.16 and 0.34 | 0.16 and 0.34 | M33 and N69 | + |
| SynIlvA1 | 0.13 and 0.43 | 0.13 and 0.43 | E26 and Q88 | + |
| SynIlvA2 | 0.13 and 0.43 | 0.13 and 0.43 | E26 and Q88 | + |
| SynFesRescue | Spectral Analysis 1 | Amino Acid/No. |
|---|---|---|
| SynFes2 | 0.13 and 0.18 | K26 and L37 |
| SynFes6 | 0.13 and 0.18 | S26 and L37 |
| SynFes1 | 0.13 | N26 |
| SynFes3 | 0.09 | Q18 |
| SynFes5 | 0.09 | Q18 |
| SynFes7 | 0.29 | Q59 |
| SynFes8 | 0.29 | Q59 |
| SynFes4 | 0.33 | M68 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štambuk, N.; Konjevoda, P. Structural and Functional Modeling of Artificial Bioactive Proteins. Information 2017, 8, 29. https://doi.org/10.3390/info8010029
Štambuk N, Konjevoda P. Structural and Functional Modeling of Artificial Bioactive Proteins. Information. 2017; 8(1):29. https://doi.org/10.3390/info8010029
Chicago/Turabian StyleŠtambuk, Nikola, and Paško Konjevoda. 2017. "Structural and Functional Modeling of Artificial Bioactive Proteins" Information 8, no. 1: 29. https://doi.org/10.3390/info8010029
APA StyleŠtambuk, N., & Konjevoda, P. (2017). Structural and Functional Modeling of Artificial Bioactive Proteins. Information, 8(1), 29. https://doi.org/10.3390/info8010029