
Minimizing Cohort Discrepancies: A Comparative Analysis of Data Normalization Approaches in Biomarker Research



Abstract
1. Introduction
2. Materials and Methods
- Normalization by total concentration:
- 2.
- Autoscaling normalization:
- 3.
- Quantile normalization:
- 4.
- Probabilistic quotient normalization:
- 5.
- Median ratio normalization:
- 6.
- Trimmed median-m value normalization:
- 7.
- Variance stabilizing normalization.
3. Results
4. Discussion
5. Limitations, Future Prospects, and Suggestions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
List of Used Abbreviations
| HIE—hypoxic–ischemic encephalopathy |
| MRN—median ration normalization |
| OPLS—orthogonal partial least squares |
| PCA—principal component analysis |
| PQN—probabilistic quotient normalization |
| TMM—trimmed mean m-value normalization |
| VSN—variance stabilizing normalization |
| VIP—variable importance projection |
References
- Badrick, T. Biological variation: Understanding why it is so important? Pract. Lab. Med. 2021, 23, e00199. [Google Scholar] [CrossRef] [PubMed]
- Higdon, R.; Kolker, E. Can “normal” protein expression ranges be estimated with high-throughput proteomics? J. Proteome Res. 2015, 14, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Chelala, L.; O’Connor, E.E.; Barker, P.B.; Zeffiro, T.A. Meta-analysis of brain metabolite differences in HIV infection. NeuroImage Clin. 2020, 28, 102436. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Siegel, L.; Zhou, J.; Zhu, M.; Tong, T.; Chen, Y.; Chu, H. Estimating the reference interval from a fixed effects meta-analysis. Res. Synth. Methods 2021, 12, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, J.; Lim, M.-S.; Seong, S.J.; Seo, J.J.; Park, S.M.; Lee, H.W.; Yoon, Y.-R. Quantile normalization approach for liquid chromatography—Mass spectrometry-based metabolomic data from healthy human volunteers. Anal. Sci. 2012, 28, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Von Heydebreck, A.; Sültmann, H.; Poustka, A.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Brix, F.; Demetrowitsch, T.; Jensen-Kroll, J.; Zacharias, H.U.; Szymczak, S.; Laudes, M.; Schreiber, S.; Schwarz, K. Evaluating the Effect of Data Merging and Postacquisition Normalization on Statistical Analysis of Untargeted High-Resolution Mass Spectrometry Based Urinary Metabolomics Data. Anal. Chem. 2024, 96, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Chua, A.E.; Pfeifer, L.D.; Sekera, E.R.; Hummon, A.B.; Desaire, H. Workflow for Evaluating Normalization Tools for Omics Data Using Supervised and Unsupervised Machine Learning. J. Am. Soc. Mass Spectrom. 2023, 34, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Shevtsova, Y.; Starodubtseva, N.; Tokareva, A.; Goryunov, K.; Sadekova, A.; Vedikhina, I.; Ivanetz, T.; Ionov, O.; Frankevich, V.; Plotnikov, E.; et al. Metabolite Biomarkers for Early Ischemic–Hypoxic Encephalopathy: An Experimental Study Using the NeoBase 2 MSMS Kit in a Rat Model. Int. J. Mol. Sci. 2024, 25, 2035. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Edwards, A.B.; Feindel, K.W.; Cross, J.L.; Anderton, R.S.; Clark, V.W.; Knuckey, N.W.; Meloni, B.P. Modification to the Rice-Vannucci perinatal hypoxic-ischaemic encephalopathy model in the P7 rat improves the reliability of cerebral infarct development after 48 hours. J. Neurosci. Methods 2017, 288, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.; Hardin, J.; Stoebel, D.M. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief. Bioinform. 2018, 19, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; von Heydebreck, A.; Sueltmann, H.; Poustka, A.; Vingron, M. Parameter estimation for the calibration and variance stabilization of microarray data. Stat. Appl. Genet. Mol. Biol. 2003, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, G.C. Variability in donor leukocyte counts confound the use of common RNA sequencing data normalization strategies in transcriptomic biomarker studies performed with whole blood. Sci. Rep. 2023, 13, 15514. [Google Scholar] [CrossRef] [PubMed]
- Abbas-Aghababazadeh, F.; Li, Q.; Fridley, B.L. Comparison of normalization approaches for gene expression studies completed with highthroughput sequencing. PLoS ONE 2018, 13, e0206312. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.; Ma, Y.; Gamagedara, S. Evaluation of statistical techniques to normalize mass spectrometry-based urinary metabolomics data. J. Pharm. Biomed. Anal. 2020, 177, 112854. [Google Scholar] [CrossRef] [PubMed]
- Dressler, F.F.; Brägelmann, J.; Reischl, M.; Perner, S. Normics: Proteomic Normalization by Variance and Data-Inherent Correlation Structure. Mol. Cell. Proteom. 2022, 21, 100269. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Kannan, S.; Chawade, A.; Bhattacharjee, A.; Govekar, R. Clinical biomarker discovery by SWATH-MS based label-free quantitative proteomics: Impact of criteria for identification of differentiators and data normalization method. J. Transl. Med. 2019, 17, 184. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Wu, D.; Zhang, J.; Pan, Y.; Kan, R.; Gao, J.; Zhou, B. Protective effect and mechanism of procyanidin B2 against hypoxic injury of cardiomyocytes. Heliyon 2023, 9, e21309. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Wang, D.; Chen, D.; Sun, Y.; Feng, X.; Shi, X.; Xu, Y.; Luo, X.; Yu, J.; Li, Y.; et al. Characterizing the effects of hypoxia on the metabolic profiles of mesenchymal stromal cells derived from three tissue sources using chemical isotope labeling liquid chromatography-mass spectrometry. Cell Tissue Res. 2020, 380, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhu, P.; Fujino, M.; Zhuang, J.; Guo, H.; Sheikh, I.; Zhao, L.; Li, X.-K. Oxidative stress in hypoxic-ischemic encephalopathy: Molecular mechanisms and therapeutic strategies. Int. J. Mol. Sci. 2016, 17, 2078. [Google Scholar] [CrossRef] [PubMed]
- Denihan, N.M.; Kirwan, J.A.; Walsh, B.H.; Dunn, W.B.; Broadhurst, D.I.; Boylan, G.B.; Murray, D.M. Untargeted metabolomic analysis and pathway discovery in perinatal asphyxia and hypoxic-ischaemic encephalopathy. J. Cereb. Blood Flow Metab. 2019, 39, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Kuligowski, J.; Solberg, R.; Sánchez-Illana, Á.; Pankratov, L.; Parra-Llorca, A.; Quintás, G.; Saugstad, O.D.; Vento, M. Plasma metabolite score correlates with Hypoxia time in a newly born piglet model for asphyxia. Redox Biol. 2017, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
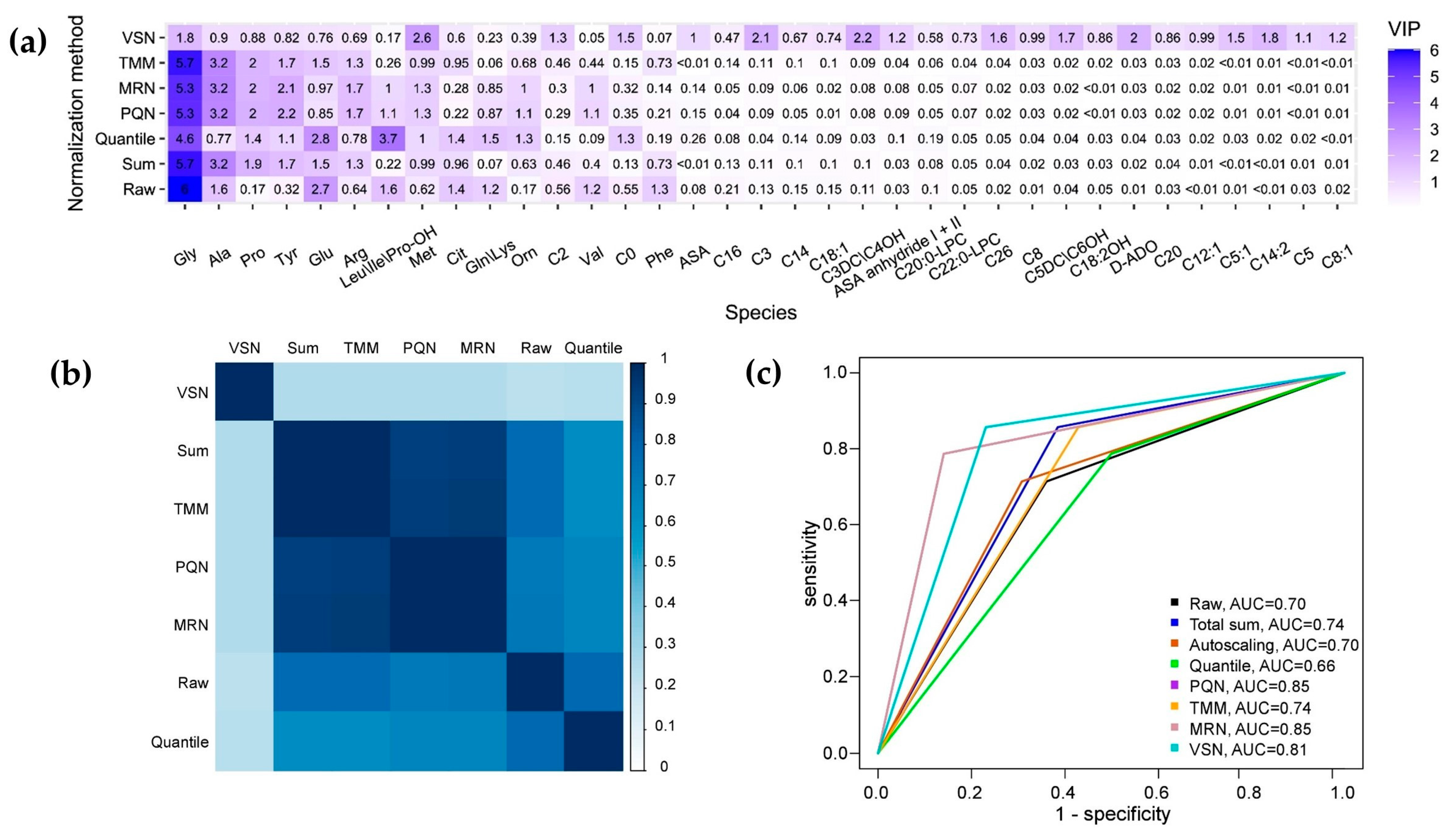
| Normalization Method | R2X | R2Y | Q2Y | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| Raw | 0.69 | 0.68 | 0.56 | 0.70 | 0.71 | 0.64 |
| Total sum | 0.76 | 0.62 | 0.47 | 0.74 | 0.86 | 0.57 |
| Autoscaling | 0.69 | 0.68 | 0.56 | 0.70 | 0.71 | 0.69 |
| Quantile normalization | 0.59 | 0.66 | 0.38 | 0.70 | 0.71 | 0.69 |
| PQN | 0.56 | 0.72 | 0.55 | 0.67 | 0.79 | 0.5 |
| MRN | 0.58 | 0.72 | 0.55 | 0.85 | 0.79 | 0.86 |
| TMM | 0.78 | 0.62 | 0.47 | 0.85 | 0.79 | 0.86 |
| VSN | 0.26 | 0.89 | 0.72 | 0.74 | 0.86 | 0.57 |
| Normalization Method | Advantages | Disadvantages |
|---|---|---|
| Total sum | The OPLS model performed well, displaying a close distribution of the biomarkers’ importance compared to the other normalized datasets (TMM, PQN, MRN) | The OPLS model’s performance is adversely affected by imbalances in the training data, resulting in a lower quality outcome compared to when the model is trained on raw data. |
| Autoscaling | None | The application of this approach does not lead to an improvement in the validation results with the test data. |
| Quantile normalization | The distribution of the biomarkers’ importance closely aligns with the distribution observed in the raw data. | The performance of the OPLS model is found to be unsatisfactory, particularly as it demonstrates sensitivity to imbalances within the training data. |
| PQN | The OPLS model performed well, displaying a close distribution of the biomarkers’ importance compared to the other normalized datasets (TMM, total sum, MRN), resulting in an enhanced validation outcome for the test data | None |
| MRN | The OPLS model performed well, displaying a close distribution of the biomarkers’ importance compared to the other normalized datasets (TMM, total sum, PQN), resulting in an enhanced validation outcome for the test data | None |
| TMM | The OPLS model performed well, displaying a close distribution of biomarkers’ importance compared to the other normalized datasets (PQN, total sum, MRN), resulting in an enhanced validation outcome for the test data | The OPLS model’s performance is adversely affected by imbalances in the training data, resulting in a lower quality outcome compared to when the model is trained on raw data. |
| VSN | The model’s exceptional quality is demonstrated by achieving the highest sensitivity during the validation on test data, indicating its robust performance and reliability in accurately predicting outcomes. | There has been a significant change in the distribution of the biomarkers’ importance, reflecting a notable shift in the key factors influencing the model’s outcomes. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokareva, A.; Starodubtseva, N.; Frankevich, V.; Silachev, D. Minimizing Cohort Discrepancies: A Comparative Analysis of Data Normalization Approaches in Biomarker Research. Computation 2024, 12, 137. https://doi.org/10.3390/computation12070137
Tokareva A, Starodubtseva N, Frankevich V, Silachev D. Minimizing Cohort Discrepancies: A Comparative Analysis of Data Normalization Approaches in Biomarker Research. Computation. 2024; 12(7):137. https://doi.org/10.3390/computation12070137
Chicago/Turabian StyleTokareva, Alisa, Natalia Starodubtseva, Vladimir Frankevich, and Denis Silachev. 2024. "Minimizing Cohort Discrepancies: A Comparative Analysis of Data Normalization Approaches in Biomarker Research" Computation 12, no. 7: 137. https://doi.org/10.3390/computation12070137
APA StyleTokareva, A., Starodubtseva, N., Frankevich, V., & Silachev, D. (2024). Minimizing Cohort Discrepancies: A Comparative Analysis of Data Normalization Approaches in Biomarker Research. Computation, 12(7), 137. https://doi.org/10.3390/computation12070137