NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
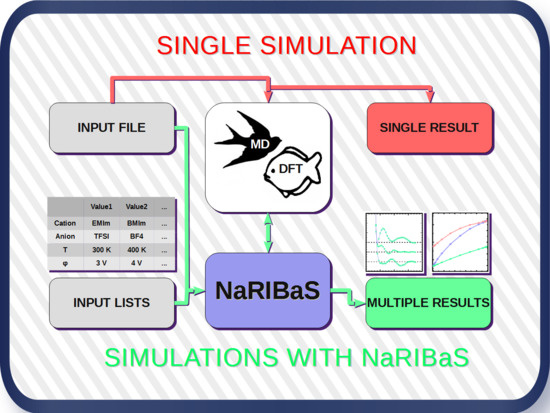
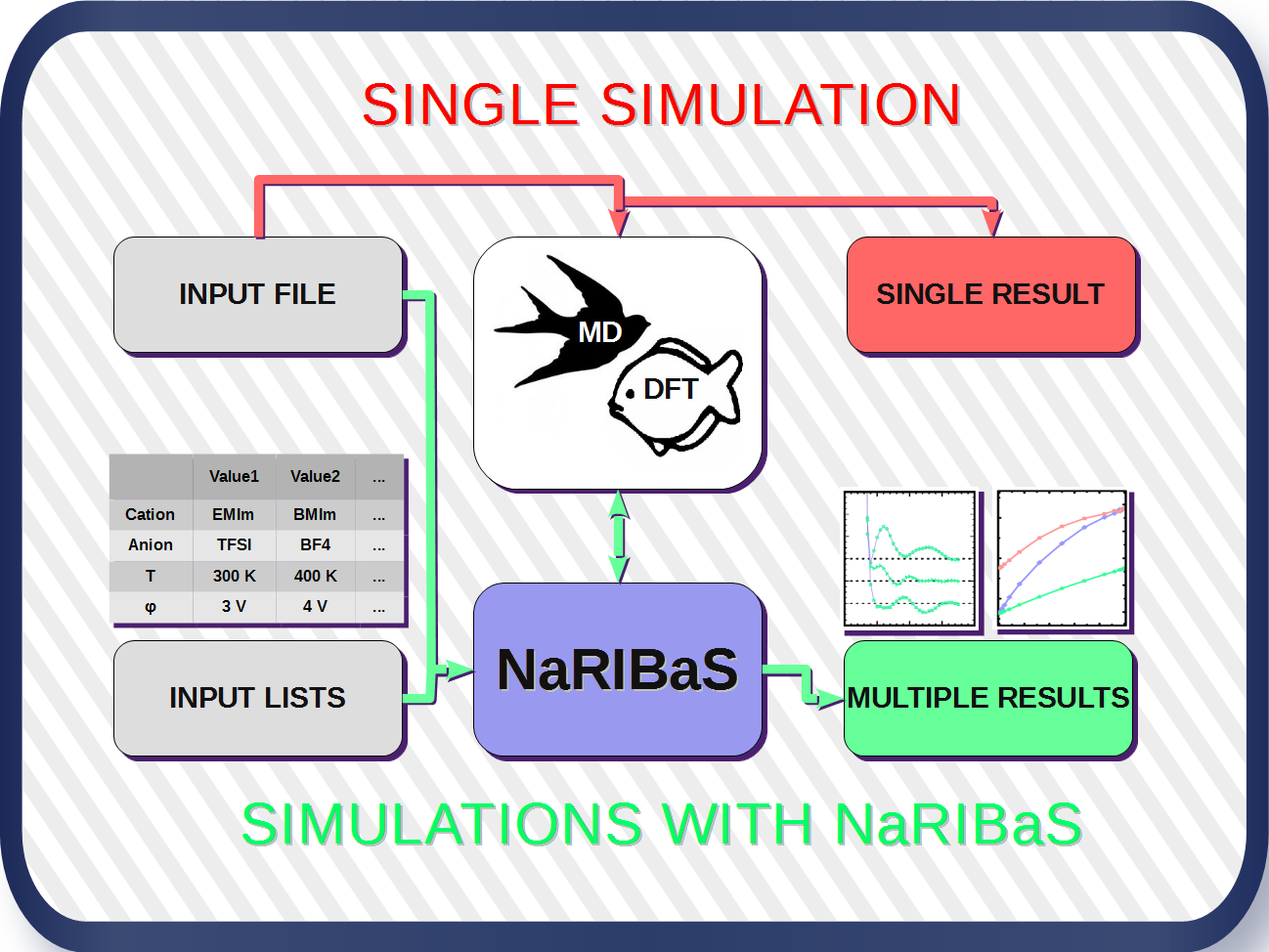
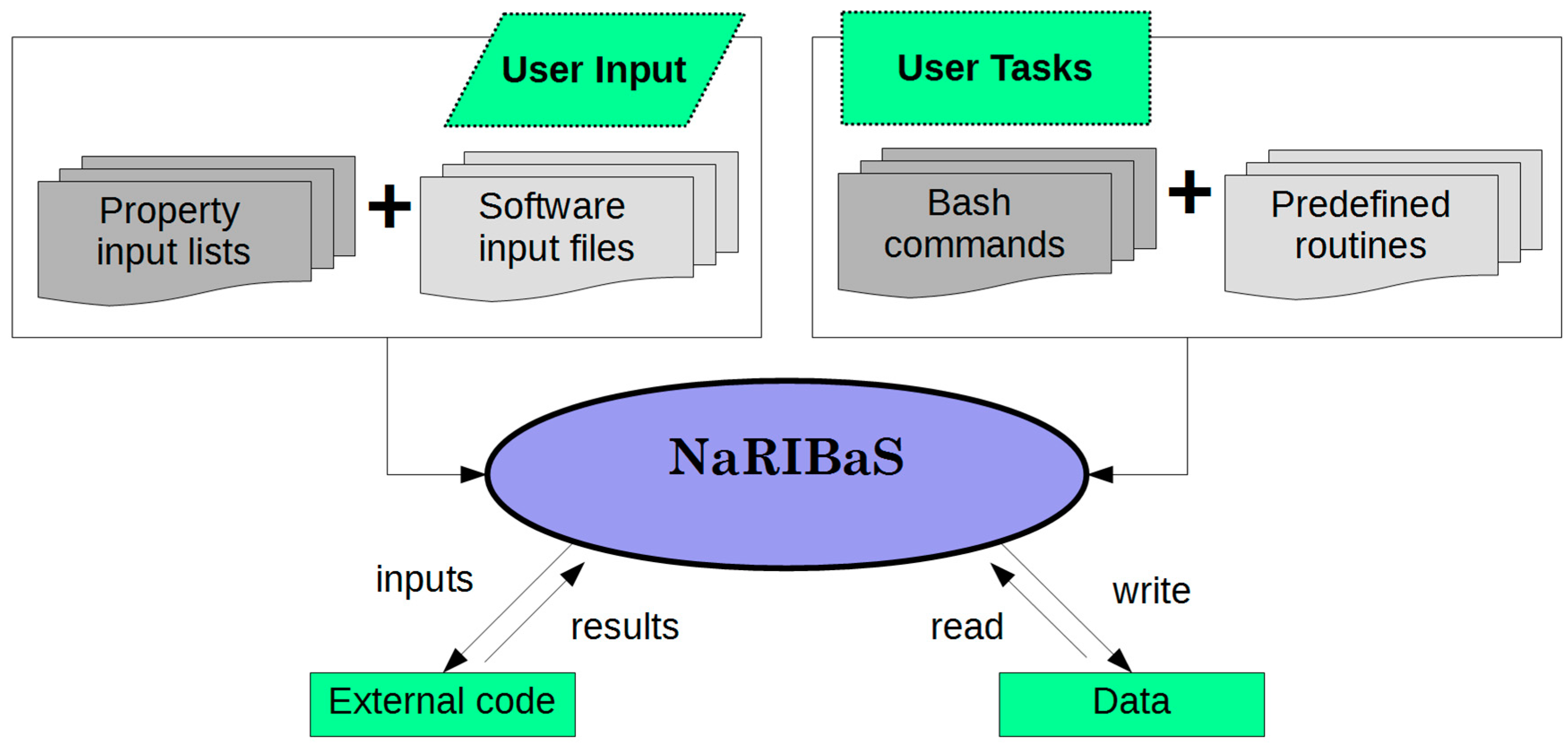
2. What is NaRIBaS?
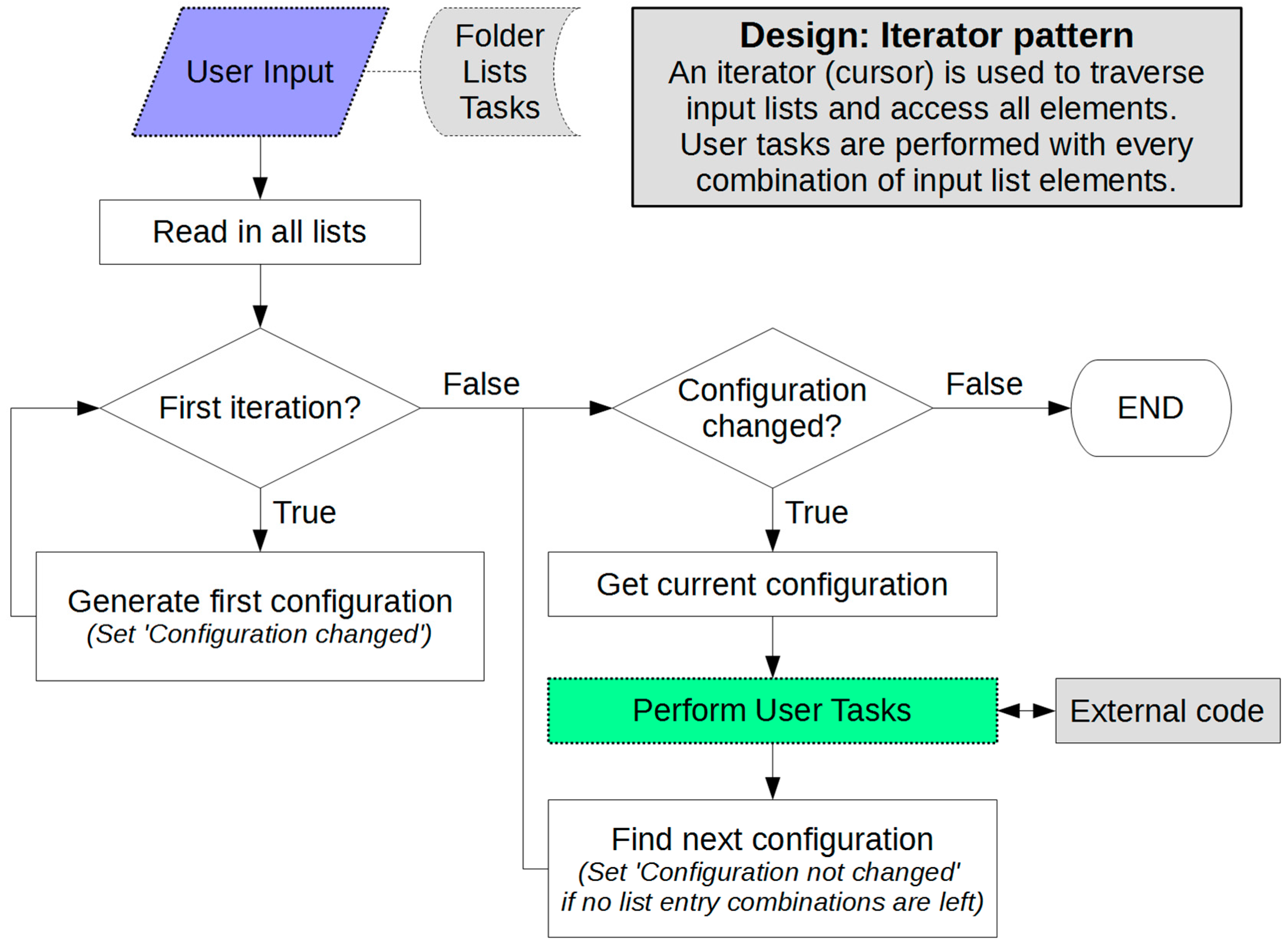
3. How Does NaRIBaS Work?
Main Features of NaRIBaS
4. NaRIBaS in Action
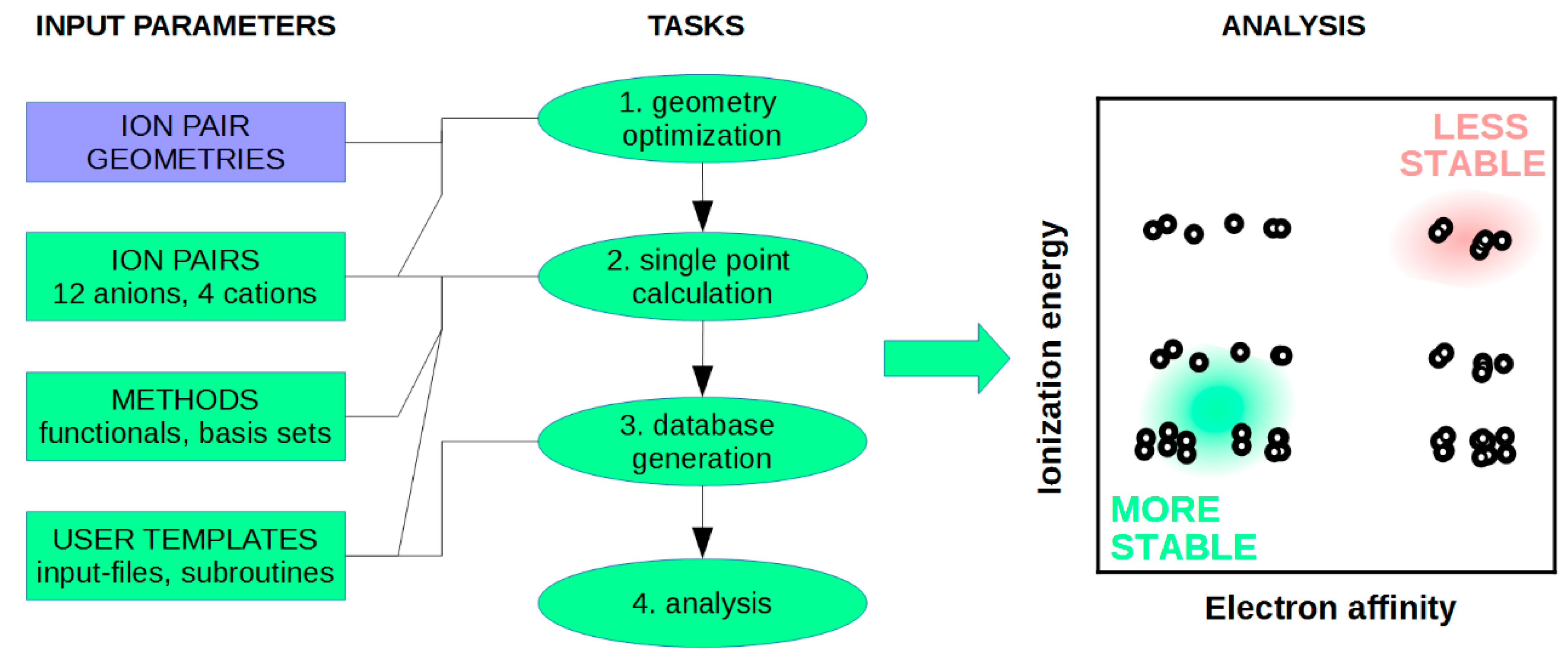
4.1. DFT Calculations
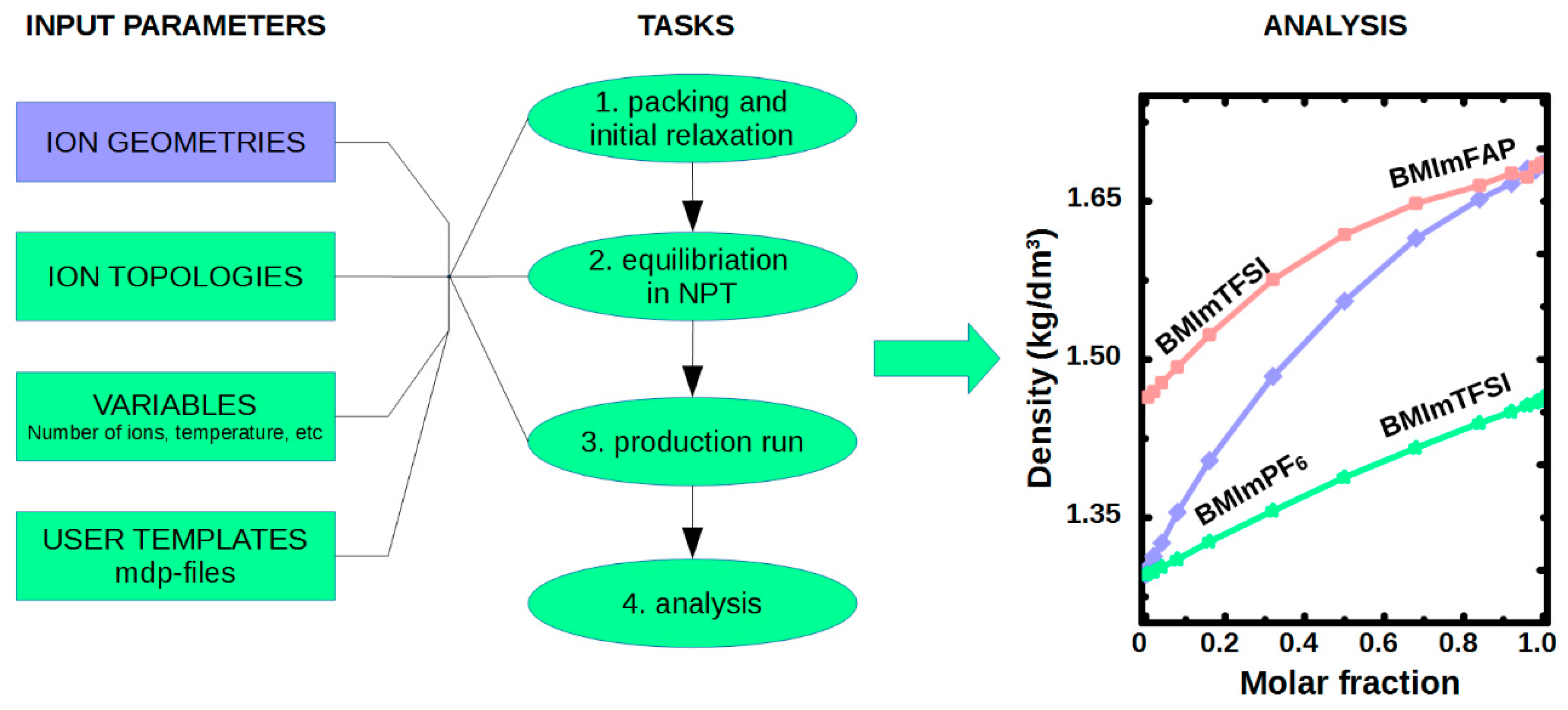
4.2. MD Simulations of Ionic Liquid in Bulk
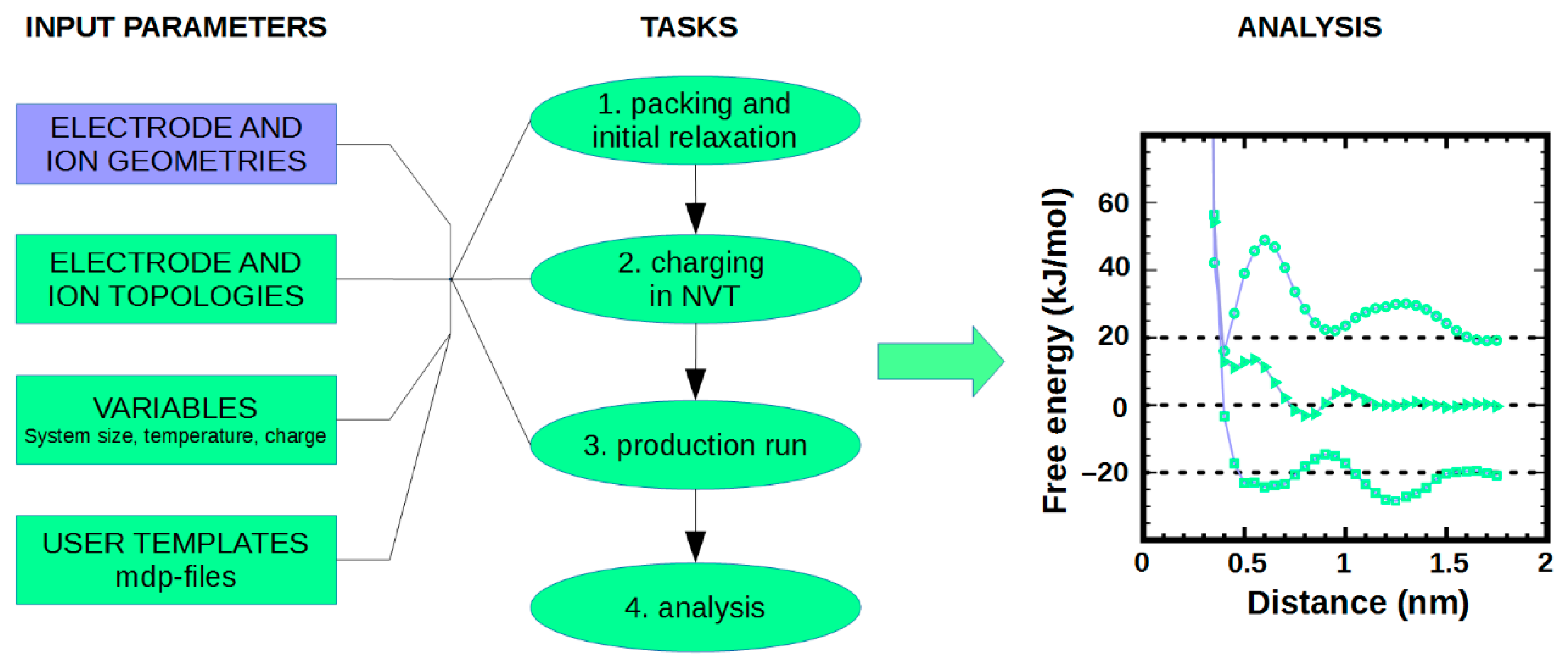
4.3. MD Simulations of Ionic Liquid at Interfaces
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fedorov, M.V.; Kornyshev, A.A. Ionic liquids at electrified interfaces. Chem. Rev. 2014, 114, 2978–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, D.R.; Tachikawa, N.; Forsyth, M.; Pringle, J.M.; Howlett, P.C.; Elliott, G.D.; Davis, J.H.; Watanabe, M.; Simon, P.; Angell, C.A. Energy applications of ionic liquids. Energy Environ. Sci. 2014, 7, 232–250. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Jain, A.; Rajput, N.N.; Cheng, L.; Zhang, Y.; Ong, S.P.; Brafman, M.; Maginn, E.; Curtiss, L.A.; Persson, K.A. The Electrolyte Genome project: A big data approach in battery materials discovery. Comput. Mater. Sci. 2015, 103, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Izgorodina, E.I.; Seeger, Z.L.; Scarborough, D.L.A.; Tan, S.Y.S. Quantum Chemical Methods for the Prediction of Energetic, Physical, and Spectroscopic Properties of Ionic Liquids. Chem. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Salanne, M. Simulations of room temperature ionic liquids: From polarizable to coarse-grained force fields. Phys. Chem. Chem. Phys. 2015, 17, 14270–14279. [Google Scholar] [CrossRef] [PubMed]
- Pirhadi, S.; Sunseri, J.; Koes, D.R. Open Source Molecular Modeling. J. Mol. Graph. Model. 2016. [Google Scholar] [CrossRef] [PubMed]
- Te Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE SC2006 Conference on High Performance Networking and Computing, Tampa, FL, USA, 11–17 November 2006; 2006; p. 43. [Google Scholar]
- Klein, C.; Sallai, J.; Jones, T.J.; Iacovella, C.R.; McCabe, C.; Cummings, P.T. A Hierarchical, Component Based Approach to Screening Properties of Soft Matter. In Foundations of Molecular Modeling and Simulation: Select Papers from FOMMS 2015; Snurr, R.Q., Adjiman, C.S., Kofke, D.A., Eds.; Molecular Modeling and Simulation; Springer: Singapore, 2016; pp. 79–92. ISBN 978-981-10-1128-3. [Google Scholar]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; Hermes, E.D.; et al. The atomic simulation environment—A Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, M.E.; Colina, C.M. pysimm: A python package for simulation of molecular systems. SoftwareX 2017, 6, 7–12. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GNU General Public License. Available online: https://www.gnu.org/copyleft/gpl.html (accessed on 4 November 2018).
- Ivaništšev, V. Repository for: NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab. GitHub Repos. 2018. Available online: https://github.com/vilab-tartu/NaRIBaS (accessed on 12 November 2018).
- Korth, M. Large-scale virtual high-throughput screening for the identification of new battery electrolyte solvents: Evaluation of electronic structure theory methods. Phys. Chem. Chem. Phys. 2014, 16, 7919–7926. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; MacFarlane, D.; Izgorodina, E.I. Assessment of Kohn-Sham Density Functional Theory and Moller-Plesset Perturbation Theory for Ionic Liquids. Phys. Chem. Chem. Phys. 2013, 15, 13664–13675. [Google Scholar] [CrossRef] [PubMed]
- Karu, K.; Ruzanov, A.; Ers, H.; Ivaništšev, V.; Lage-Estebanez, I.; García de la Vega, J.M. Predictions of Physicochemical Properties of Ionic Liquids with DFT. Computation 2016, 4, 25. [Google Scholar] [CrossRef]
- Karu, K.; Mišin, M.; Ers, H.; Sun, J.; Ivaništšev, V. Performance of SCAN density functional for a set of ionic liquid ion pairs. Int. J. Quantum Chem. 2018, 118, e25582. [Google Scholar] [CrossRef] [Green Version]
- Lage-Estebanez, I.; Ruzanov, A.; de la Vega, J.M.G.; Fedorov, M.V.; Ivaništšev, V.B. Self-interaction error in DFT-based modelling of ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 2175–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Karu, K.; Ers, H.; Mišin, M.; Sun, J.; Ivanistsev, V. Data for the article “Performance of SCAN density functional method for a set of ionic liquids”. GitHub Repos. 2017. [Google Scholar] [CrossRef]
- Voroshylova, I.V.; Ferreira, E.S.C.; Malček, M.; Costa, R.; Pereira, C.M.; Cordeiro, M.N.D.S. Influence of the anion on the properties of ionic liquid mixtures: A molecular dynamics study. Phys. Chem. Chem. Phys. 2018, 20, 14899–14918. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to molecular simulation. Mol. Simul. 2002, 28, 385–471. [Google Scholar] [CrossRef]
- Ivaništšev, V. Gromacs topology files for common Ionic Liquids. GitHub Repos. 2018. [Google Scholar] [CrossRef]
- Merlet, C.; Limmer, D.T.; Salanne, M.; van Roij, R.; Madden, P.A.; Chandler, D.; Rotenberg, B. The Electric Double Layer Has a Life of Its Own. J. Phys. Chem. C 2014, 118, 18291–18298. [Google Scholar] [CrossRef] [Green Version]
- Rotenberg, B.; Salanne, M. Structural Transitions at Ionic Liquid Interfaces. J. Phys. Chem. Lett. 2015, 6, 4978–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.L.; Meng, Q.; Fan, J. Charge driven lateral structural evolution of ions in electric double layer capacitors strongly correlates with differential capacitance. Phys. Chem. Chem. Phys. 2018, 20, 8054–8063. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Vatamanu, J.; Borodin, O.; Bedrov, D. A molecular dynamics simulation study of the electric double layer and capacitance of [BMIM][PF6] and [BMIM][BF4] room temperature ionic liquids near charged surfaces. Phys. Chem. Chem. Phys. 2013, 15, 14234–14247. [Google Scholar] [CrossRef] [PubMed]
- Breitsprecher, K.; Szuttor, K.; Holm, C. Electrode Models for Ionic Liquid-Based Capacitors. J. Phys. Chem. C 2015, 119, 22445–22451. [Google Scholar] [CrossRef]
- Chen, M.; Goodwin, Z.A.H.; Feng, G.; Kornyshev, A.A. On the temperature dependence of the double layer capacitance of ionic liquids. J. Electroanal. Chem. 2017. [Google Scholar] [CrossRef]
- Docampo-Álvarez, B.; Gómez-González, V.; Montes-Campos, H.; Otero-Mato, J.M.; Méndez-Morales, T.; Cabeza, O.; Gallego, L.J.; Lynden-Bell, R.M.; Ivaništšev, V.B.; Fedorov, M.V.; et al. Molecular dynamics simulation of the behaviour of water in nano-confined ionic liquid–water mixtures. J. Phys. Condens. Matter 2016, 28, 464001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlet, C.; Péan, C.; Rotenberg, B.; Madden, P.A.; Daffos, B.; Taberna, P.-L.; Simon, P.; Salanne, M. Highly confined ions store charge more efficiently in supercapacitors. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salanne, M. Ionic Liquids for Supercapacitor Applications. Top. Curr. Chem. 2017, 375. [Google Scholar] [CrossRef]
- Ivaništšev, V.; Fedorov, M.V.; Lynden-Bell, R.M. Screening of Ion–Graphene Electrode Interactions by Ionic Liquids: The Effects of Liquid Structure. J. Phys. Chem. C 2014, 118, 5841–5847. [Google Scholar] [CrossRef]
- Ivaništšev, V.; Méndez-Morales, T.; Lynden-Bell, R.M.; Cabeza, O.; Gallego, L.J.; Varela, L.M.; Fedorov, M.V. Molecular origin of high free energy barriers for alkali metal ion transfer through ionic liquid–graphene electrode interfaces. Phys. Chem. Chem. Phys. 2016, 18, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Gómez-González, V.; Docampo-Álvarez, B.; Méndez-Morales, T.; Cabeza, O.; Ivaništšev, V.B.; Fedorov, M.V.; Gallego, L.J.; Varela, L.M. Molecular dynamics simulation of the structure and interfacial free energy barriers of mixtures of ionic liquids and divalent salts near a graphene wall. Phys. Chem. Chem. Phys. 2017, 19, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Ruzanov, A.; Lembinen, M.; Jakovits, P.; Srirama, S.N.; Voroshylova, I.V.; Cordeiro, M.N.D.S.; Pereira, C.M.; Rossmeisl, J.; Ivaništšev, V.B. On the thickness of the double layer in ionic liquids. Phys. Chem. Chem. Phys. 2018, 20, 10275–10285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchner, K.; Kirchner, T.; Ivaništšev, V.; Fedorov, M.V. Electrical double layer in ionic liquids: Structural transitions from multilayer to monolayer structure at the interface. Electrochimica Acta 2013, 110, 762–771. [Google Scholar] [CrossRef]
- Ivaništšev, V.; Kirchner, K.; Kirchner, T.; Fedorov, M.V. Restructuring of the electrical double layer in ionic liquids upon charging. J. Phys. Condens. Matter 2015, 27, 102101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivaništšev, V.; Fedorov, M.V. Interfaces between Charged Surfaces and Ionic Liquids: Insights from Molecular Simulations. Electrochem. Soc. Interface 2014, 23, 65–69. [Google Scholar] [CrossRef] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roos Nerut, E.; Karu, K.; Voroshylova, I.V.; Kirchner, K.; Kirchner, T.; Fedorov, M.V.; Ivaništšev, V.B. NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab. Computation 2018, 6, 57. https://doi.org/10.3390/computation6040057
Roos Nerut E, Karu K, Voroshylova IV, Kirchner K, Kirchner T, Fedorov MV, Ivaništšev VB. NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab. Computation. 2018; 6(4):57. https://doi.org/10.3390/computation6040057
Chicago/Turabian StyleRoos Nerut, Eva, Karl Karu, Iuliia V. Voroshylova, Kathleen Kirchner, Tom Kirchner, Maxim V. Fedorov, and Vladislav B. Ivaništšev. 2018. "NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab" Computation 6, no. 4: 57. https://doi.org/10.3390/computation6040057
APA StyleRoos Nerut, E., Karu, K., Voroshylova, I. V., Kirchner, K., Kirchner, T., Fedorov, M. V., & Ivaništšev, V. B. (2018). NaRIBaS—A Scripting Framework for Computational Modeling of Nanomaterials and Room Temperature Ionic Liquids in Bulk and Slab. Computation, 6(4), 57. https://doi.org/10.3390/computation6040057