1. Introduction
Despite the initial great promise of cell therapy, clinical trials have come up in many cases with conflicting results and controversial clinical outcomes [
1,
2,
3]. Known issues for the success of the treatment are linked to the delivery of a stable and viable population of stem cells at the damaged site, that is in turn strongly dependent on the administration route and dose. The abrupt change of microenvironment conditions after grafting cells expanded in vitro into the host may further contribute to cell loss. Finally, the host immune response is often indicated as a major cause of graft rejection.
Several strategies have been proposed to improve the delivery and retention of a stable and dense population of therapeutic cells at the damaged site. Amongst these, the main strategy relies upon either cell (micro)encapsulation and/or cell scaffolding [
4,
5,
6,
7]. Cell encapsulation makes use of biocompatible hydrogels (e.g., alginate, hyaluronic acid, gelatin/collagen) to create a 3D environment in which cells can be embedded. Cell encapsulation is often intended to shield therapeutic cells from the host hostile environment by preventing the infiltration of immune cells. As an example, embedding of human mesenchymal stromal cells (hMSCs) in a gelatin matrix prior to surgical grafting into the myocardium in swine proved to be critical for increasing intra-myocardial retention of hMSCs [
8]. Cell scaffolds are made by synthetic biocompatible materials such as poly(lactide-co-glycolide), PLGA, bioengineered to provide a solid structural support to implanted cells and to mimic the extracellular matrix, allowing cells to proliferate and/or differentiate in the desired way [
9]. Seeding neuronal stem cells on solid supports (i.e., the cell scaffolds) prior to implantation has been shown to be essential to elicit tissue regeneration in brain repair after stroke [
6]. Solid scaffolds can be fabricated in different size and forms (including solid foams, nanofibers, microspheres, or microtubules), depending on their specific application [
10]. Very interestingly, scaffolds can be designed to release bioactive molecules, such as growth factors, DNA, or drugs, in a controlled manner to facilitate tissue regeneration [
11].
The variability among individuals in regards to graft immune rejection still remains an issue [
12]. Immune response can be directed against cells, against their solid supports or encapsulating materials, triggered by the surgical act, or a combination of them. Foreign Body Response (FBR) has been shown to cause fibrosis through activation of macrophages and dendritic cells as a reaction to alginate capsules [
13]. In the absence of methods enabling the real time-monitoring of the integrity of the cell population and of their function, it is therefore very hard to assess whether inconsistent endpoint clinical outcomes are due to graft rejection or poor intrinsic efficacy of the therapy. To bridge the knowledge gap between cell therapy administration and endpoint health outcomes, there is a need for non-invasive, clinically compliant methods enabling to follow-up longitudinally the cell state [
14]. Magnetic Resonance Imaging (MRI) has gained a prominent role in the field of cell therapy because it is minimally invasive, can image tissues at any depth in the body, and offer an acceptable compromise between time/space resolution and sensitivity. Cell tracking by MRI allows one to follow the migration and homing of cells after introduction within the host body [
14,
15]. An essential requirement to track stem cells by MRI is the incorporation of an imaging probe (also called the contrast agent, CA) into the cells of interest in order to distinguish them from their surroundings (direct labelling). Superparamagnetic Iron Oxide (SPIO) nanoparticles have been extensively used to track cells by MRI. Graft rejection can be assessed by following the homing of intravenously administered SPIO-labeled macrophages in kidney transplantation [
16] and xenotransplantation models [
17]. Impairment of SPIO-labelled pancreatic islet allografts due to rejection can be predicted in advance of functional failure by MRI [
18]. There are, however, drawbacks with the use of SPIO-labelled cells linked to the long-term persistence of SPIO signal after the death of transplanted cells, leading to false results [
19,
20]. Furthermore, there are reports on the detrimental effects on stem cell multipotentiality linked to the high payload of intracellular labels [
21].
As an alternative to direct cell labelling, labelling of the extracellular matrix within cell capsules was proposed, under the assumption that the cell state can be inferred by assessing the capacity of the extracellular matrix (ECM) microenvironment to support cell survival [
22,
23]. Chan et al. reported a pH-nanosensor-based MRI probe that could monitor microenvironment pH within cell-loaded microcapsules in vivo and non-invasively. Onset of acidosis was shown to report about cell death within grafted capsules [
24]. In another study, cell infiltration and FBR response to alginate capsules could be detected based on the Magnetization Transfer MR properties of the ECM microenvironment [
13]. We recently presented silica microspheres decorated with Gd-complexes as a redox-responsive MRI probe to follow-up hypoxia within cell-embedding hydrogels [
25]. In principle, a variety of microenvironment features can be probed, providing that a suitable responsive system is available (for instance, liposomes grafted with MRI-labelled substrates for matrix metalloproteinases can be used to label the extracellular microenvironment for probing matrix remodeling) [
26].
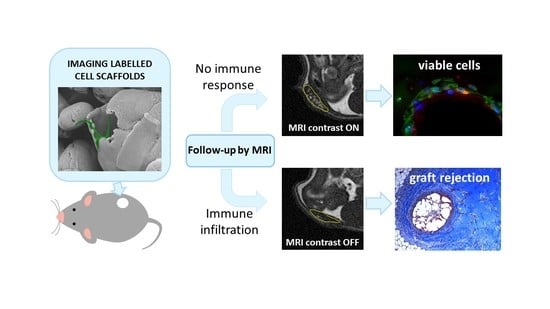
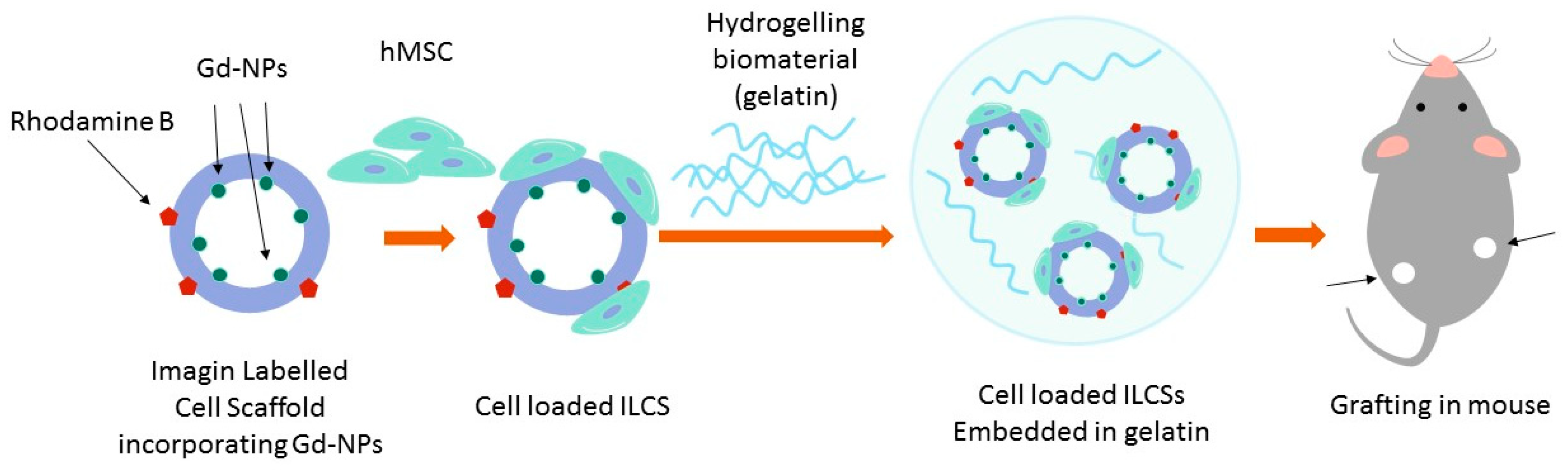
In this study, we present novel biocompatible microparticles integrating a cell scaffolding function for hMSCs with an imaging function to monitor immune response by MRI. Such Imaging Labelled Cell Scaffolds (ILCSs) are designed to be seeded with cells, then embedded into a gelatin matrix, and finally grafted into the host (
Scheme 1). ILCSs are based on microparticles composed of poly(lactide-co-glycolide) (PLGA) and chitosan (CHT). These microparticles incorporate gadolinium fluoride nanoparticles (Gd-NPs) as the T
1 MRI contrast agent [
27]. As such, the incorporated Gd-NPs are MRI silent, as they are shielded from any interaction with bulk water molecules. However, Gd-NPs are slowly released into the extracellular milieu, where they become activated for T
1-dependent MRI contrast enhancement (
Scheme 2). Due to their size (hydrodynamic diameter of ca. 12 nm; inorganic core 2 nm) [
27] and interactions with the macromolecular components of the extracellular matrix, diffusion of Gd-NPs within the hydrogel is restricted, and local activation of MRI contrast is expected. Foreign body response against scaffolded cells is expected to lead to fibrosis, encapsulation of ILCSs, and scavenging of released Gd-NPs by phagocytic cells, preventing ultimately the activation contrast enhancement.
3. Discussion
The delivery of therapeutic cells in association with biomaterials provides the outstanding opportunity to add microenvironment-responsive imaging labels in a form interspersed with therapeutic cells. While the incorporation of MRI probes into the extracellular space within hydrogel-based microcapsules has several examples [
22], the labelling of cell scaffolds received comparatively much less attention [
28,
29]. To the best of our knowledge, this is the first study with cell scaffolds labelled with MRI probes endowed with responsivity to the cell extracellular microenvironment. The system we designed is intended to detect the infiltration of host cells in the gelatin matrix, embedding scaffolded cells. It is worth emphasizing that the matrigel we used for cell embedding does not provide immune-isolation in itself. As opposite, it is permeable to host cells and allows for matrix remodelling. It was chosen for a better delivery of cell grafts, and to model cell infiltration within the graft. If immunoisolation of embedded cells is sought for a specific implementation of cell therapy, microencapsulation through suitable hydrogelling polymers, such as alginate, must be used instead.
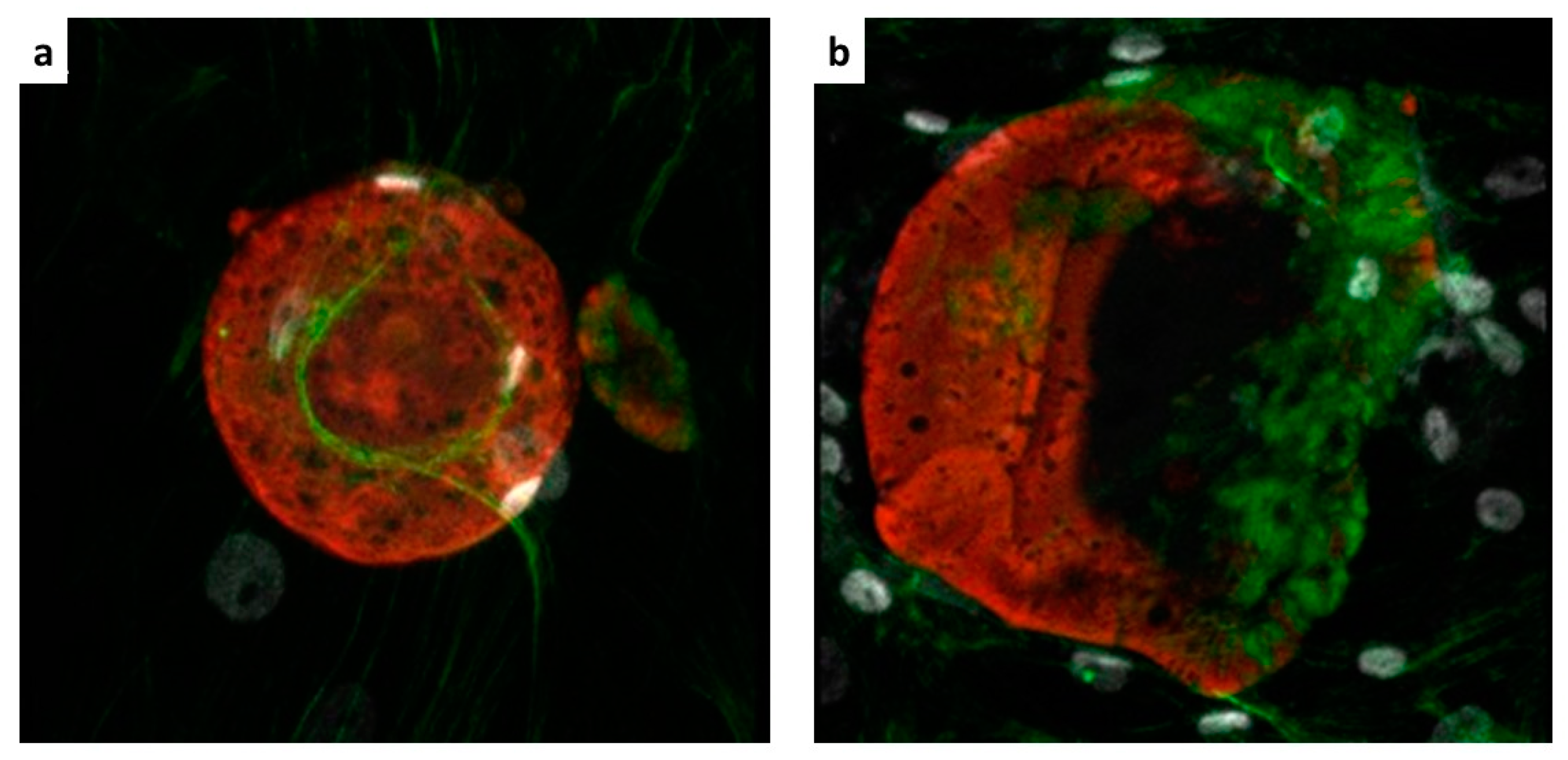
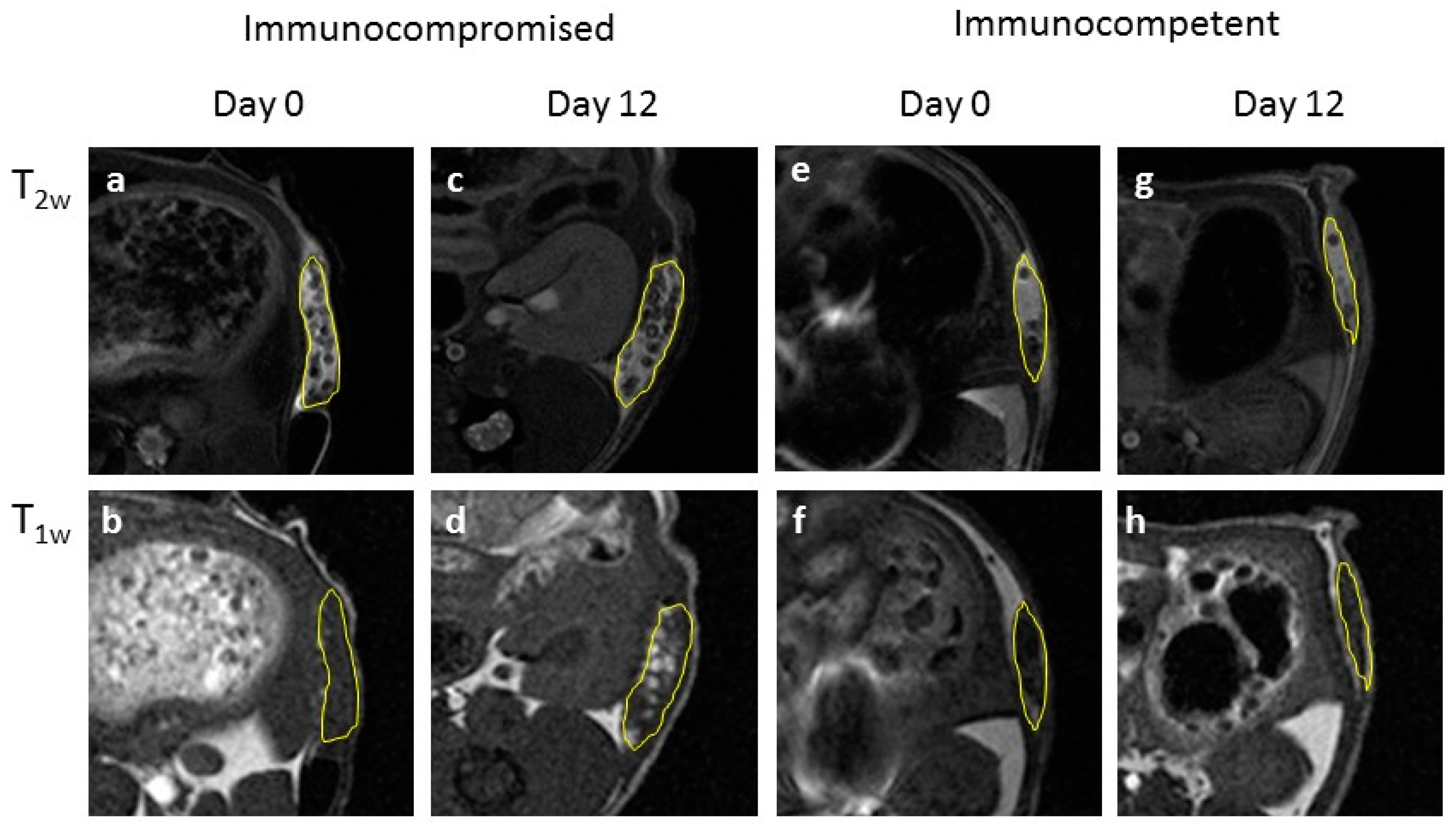
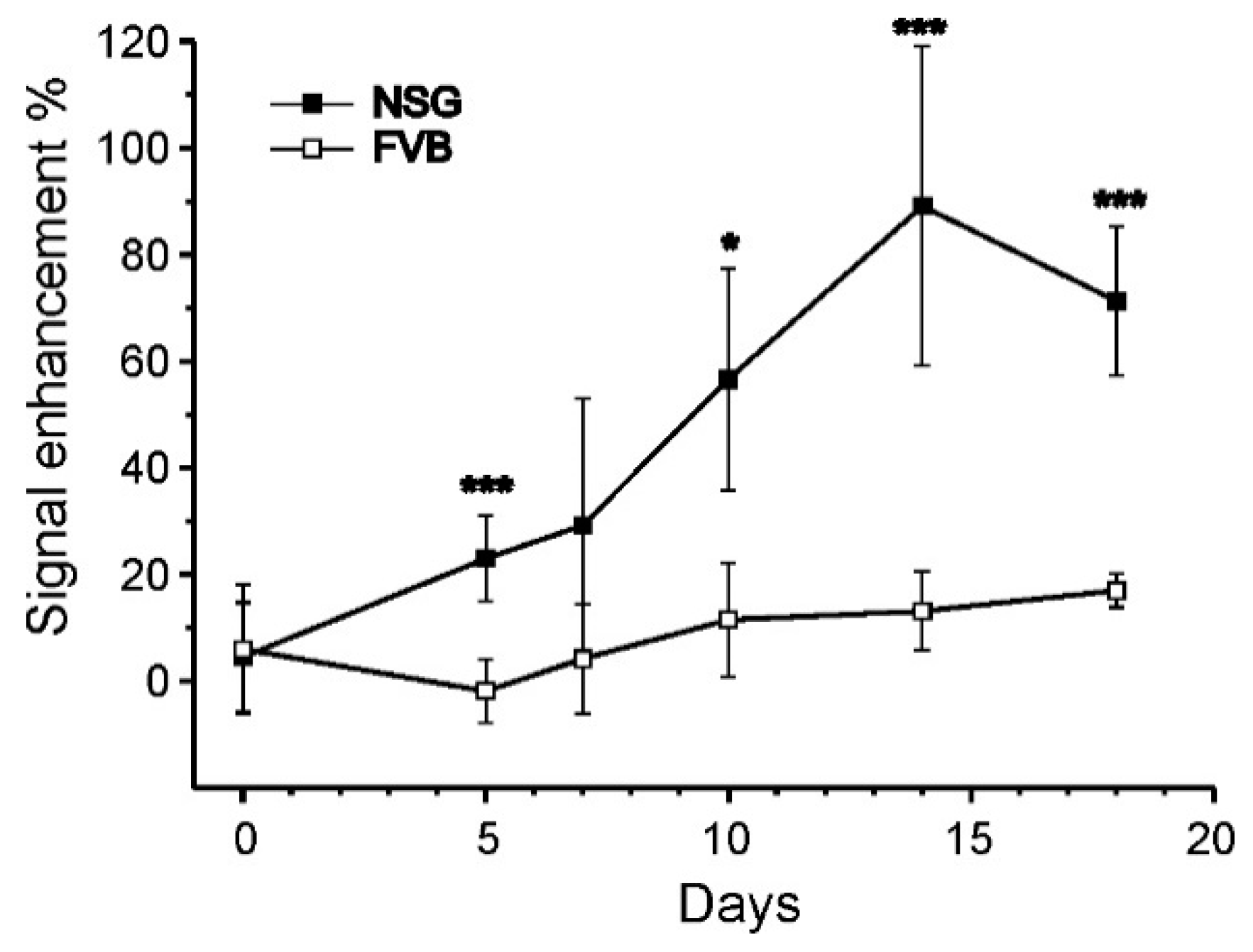
We performed an initial evaluation of our follow-up system on either immunocompromised or immunocompetent mice, under the hypothesis of no immune response in the former animals, and occurrence of immune response in the latter. Endpoint ex vivo histology confirmed such an hypothesis, as we observed poor or no infiltration of host cells with immunocompromised mice, and extensive infiltration and matrix remodelling in immunocompetent mice. The extent of cell infiltration was not affected by the presence of hMSCs, as no significant histologic differences between +hMSCs and –hMSCs cell grafts were found. The endpoint histologic observation was nicely paralleled by MRI observations. We detected a progressive appearance of MRI contrast in grafts devoid of host infiltrate. On the other hand, poor MRI contrast appeared in the presence of appreciable infiltration, likely due to the encapsulation of ILCSs within fibrotic capsules and to the scavenging of released Gd-NPs by phagocytic cells. Therefore, the imaging response well correlates with cell infiltration within the matrigel phase.
The relationship between the MRI response and cell survival is less clear. Surviving hMSCs were detected ex vivo both in infiltrated and non-infiltrated grafts. The survival of xenogeneic cells up to 20-days post transplantation even under the circumstance of strong infiltration can be due to the type of cells we have used in our work. A number of studies have shown that hMSCs have unique immunoregulatory functions [
30,
31,
32] that make them promising for long-term cell therapy. Human MSCs were xenografted in rat and swine without immunosuppressant treatment and gene expression was imaged by PET [
8]. Human MSCs were xenotransplanted in intervertebral discs in a xenogeneic porcine model and shown to survive for months [
33]. Mouse MSCs showed unique immunologic tolerance, allowing their engraftment into a xenogeneic environment [
34]. As +hMSC implants were indistinguishable from –hMSC implants, it can be speculated that immune response is directed against the microparticles rather than against xenogeneic cells. This would be in line with other reports, showing a mild FBR directed against alginate microcapsules [
13]. In summary, while MR image response is clearly related to host cell infiltration, it cannot be unambiguously related to the death of transplanted cells. Further quantitative assessment of survival of hMSCs cells within in vivo xenograft is needed to shed light on this matter.
4. Materials and Methods
4.1. Synthesis and Characterization of ILCSs
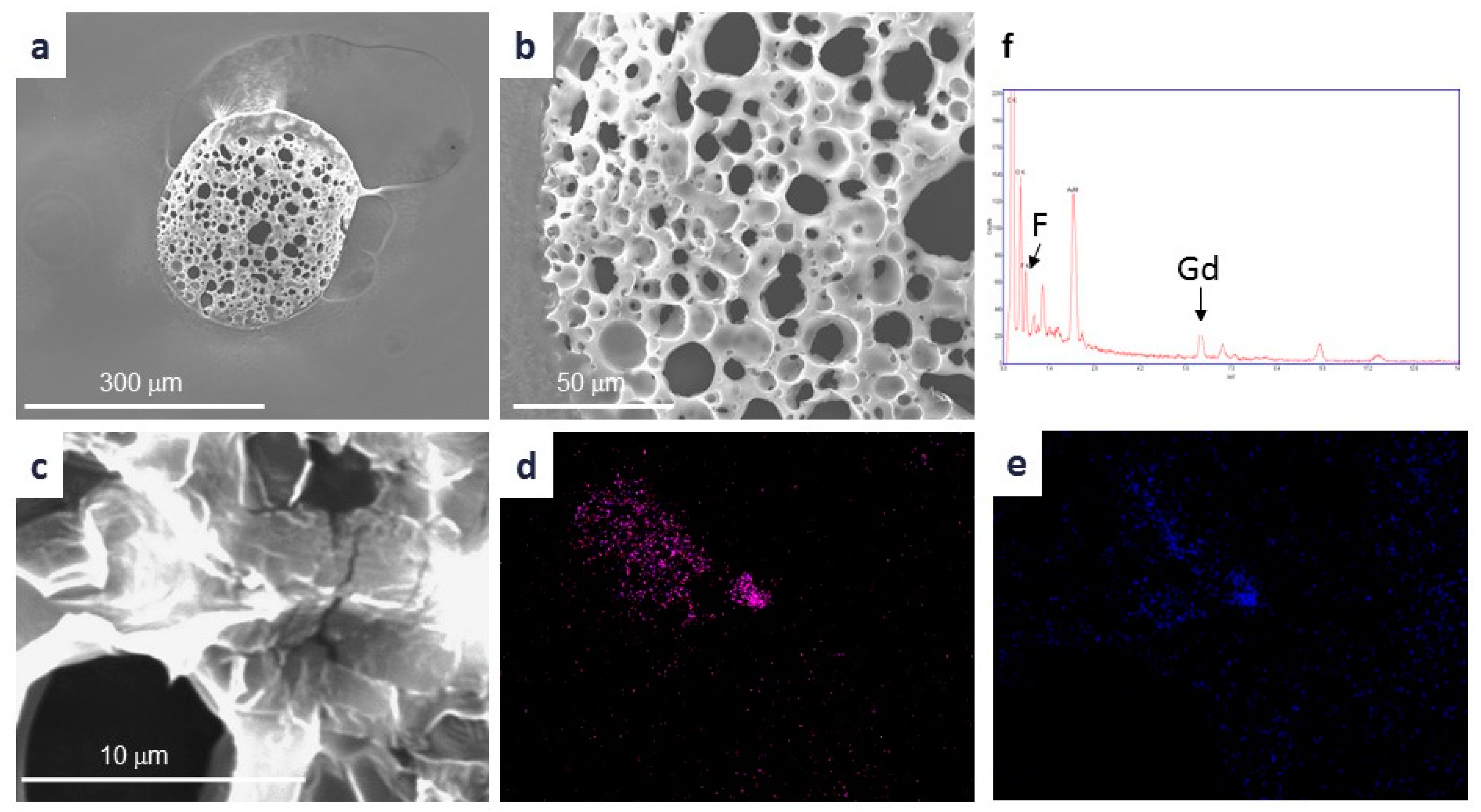
Citrate-coated GdF
3 nanoparticles (Gd-NPs) were prepared as described previously [
27]. ILCSs incorporating Gd-NPs were prepared by a modified double emulsion-solvent evaporation technique [
35]. Firstly, one gram of commercial PLGA granules (Durect Lactel Absorbable Polymers, Cupertino, CA, USA; lactide-to-glycolide ratio 50:50; Mw 40,000–50,000 g/mol) was dissolved into 5 mL of chloroform for 24 h (solution A). Solution B was prepared by dissolving 100 mg of PVA (Sigma-Aldrich, St. Louis, MO, USA, Mowiol™ 4–88, Mw ~31,000) in 100 mL of dd-H
2O on a magnetic stirrer at 1200 rpm and 90 °C for 30 min. After that, solution B was left to cool down at room temperature. Meanwhile, for the preparation of solution C, 50 mg of CHT (Sigma-Aldrich, medium molecular weight, 75–85% de-acetylated) were dissolved in 50 mL of 1% (
v/
v) acetic acid by stirring for 30 min at 500 rpm. The pH of solution C was adjusted to pH 5 with NaOH, and solution C was added to solution B under stirring at 1200 rpm for 60 min (solution D). The primary emulsion was prepared by dropwise addition of 1 mL Gd-NPs suspension (20 mM gadolinium concentration) into solution A, under 5 min homogenization using a high-energy homogenizer (Ultra-Turrax T25, IKA, Königswinter, Germany). This PLGA/Gd-NPs emulsion was added dropwise to the mixture of CHT and PVA (i.e., solution D), under stirring at 1200 rpm, which led to the formation of a double emulsion. The final emulsion was left under stirring for 20 h to promote chloroform evaporation. The microparticles formed within the final stage were recovered by filtration and washed 3 times with dd-H
2O on filter paper and finally allowed to dry in air at RT. Control microparticles (i.e., gadolinium-free) were prepared by the same protocol but in the absence of Gd-NPs.
The microparticles were labelled with rhodamine B to allow for optical imaging. One hundred mg of microspheres were placed in a tube with 2.5 mL of HEPES buffer (3.8 mM HEPES, 150 mM NaCl, pH 7.4). 1.2 mg of rhodamine B isothiocyanate (Sigma-Aldrich) were dissolved in 1 mL of HEPES buffer. 500 μL of the fluorescent tag solution were added to the microspheres and the reaction mixture kept under shaking at room temperature for 24 h. 3.5 mL of HEPES buffer were added to the solution and the microparticles were washed 3 times for 15 min. The micropaticles were dialyzed for 48 h against HEPES. The recovered microspheres were finally recovered by filtration and allowed to dry in air.
The gadolinium content of microparticles was determined by means Inductively Coupled Plasma-Mass Spectrometry, ICP-MS (Spectro Genesis ICP-OES, Spectro Analytical Instruments, Kleve, Germany) equipped with a crossflow nebulizer. Plasma was generated by argon with a power of 1400 W. Flow conditions, coolant flow 12.00 L/min, auxiliary flow 0.60 L/min, and nebulizer flow 1.00 L/min. Determinations were done in triplicate. Prior to the analysis, the sample was mineralized in HNO3/HCl 1:3 v/v (aqua regia; 5 mL) and HF (2 mL) at 100 °C for 24 h.
Scanning Electron Microscopy (SEM) micrographs were taken with a Leo 1550 SEM instrument (Zeiss, Oberkochen, Germany). Samples were mounted on aluminum stubs using a double-sided adhesive carbontape and sputtered with Au/Pd with a plasma current of 30 mA for 30 s. The thickness of Au/Pd layer was approximately 10 nm.
Optical images were acquired using an Olympus BX41 optical microscope (Olympus Corporation, Tokyo, Japan).
4.2. Cell Culture and Biocompatibility Assessment
Human MSCs were purchased from Lonza, cultured and characterized, as previously described [
36,
37,
38]. hMSCs were maintained in the presence of cysteine free Mesenchymal Stem Cells Basal Medium (MSCBM, Lonza, Basel, Switzerland). All cell preparations at different passages of culture expressed the typical MSC markers, CD105, CD73, CD44, CD90, and CD146 and not hematopoietic markers like CD45, CD14 and CD34, as evaluated by flow cytometry assay [
39] (see
Table S1 for the complete list of antibodies used in this study).
4.2.1. hMSC Culture on ILCSs
The culture of hMSCs on ILCSs was performed by incubating six mg of ILCSs (containing approx. 110 nmol of gadolinium under the form of Gd-NPs) with 1.5 × 104 hMSCs in the normal culture medium. hMSCs and ILCSs were maintained in incubator at 37 °C for 4 days; after that, further 1.5 × 104 hMSCs were added to the flasks on the ILCSs and maintained in culture for up to 20 days.
4.2.2. Biocompatibility Assessment
The biocompatibility of the microspheres, incubated with hMSCs, has been assessed for up to 20 days by evaluating cell viability, morphology, flow cytometry analysis, and in vitro osteogenic and adipogenic differentiation.
MTT assay. For the evaluation of hMSCs vitality on ILCSs, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed. Briefly, ILCSs after 7 days of culture were re-suspended in 300 μL of PBS solution for efficient disaggregation. ILCS-derived cells were plated in 96-well plates in triplicates and analyzed according to the manufacturer’s instructions (Merck-Millipore, Burlington, MA, USA).
Flow cytometry assay. hMSCs were detached from ILCSs with non-enzymatic cell dissociation solution (Sigma, St. Louis, MO, USA), washed in PBS and then incubated for 30 min at 4 °C with the appropriate fluorescein isothiocyanate (FITC) or phycoerythrin (PE) conjugate Abs or with the irrelevant control in PBS containing 0.1 BSA (Sigma). The following antibodies, all PE- or FITC-conjugated, were used, anti-CD146, -CD105, -CD90, CD73, CD44, alpha 5 integrin. Cells were analysed on a FACS (Becton Dickinson, Franklin Lakes, NJ, USA). Ten thousand cells were analysed in each experimental point.
In Vitro Osteogenic and Adipogenic differentiation. For adipogenic differentiation, hMSCs were recovered after incubation with ILCSs and incubated in appropriated adipogenic differentiation medium (Lonza) [
39]. The medium was changed two times per week for 3 weeks. The cells were fixed with 10% formalin for 20 min at room temperature and stained with 0.5% Oil Red O (Sigma) in methanol for 20 min at room temperature. For osteogenic differentiation, hMSCs were incubated in osteogenic differentiation medium (Lonza) [
39]. The media was changed two times per week for 3 weeks. Then cells were fixed with 10% formalin for 20 min at room temperature and stained with Alizarin Red, pH 4.1 (Sigma) for 20 min at room temperature. Images of undifferentiated or differentiated cells were acquired at 20× magnification.
4.3. Confocal Imaging
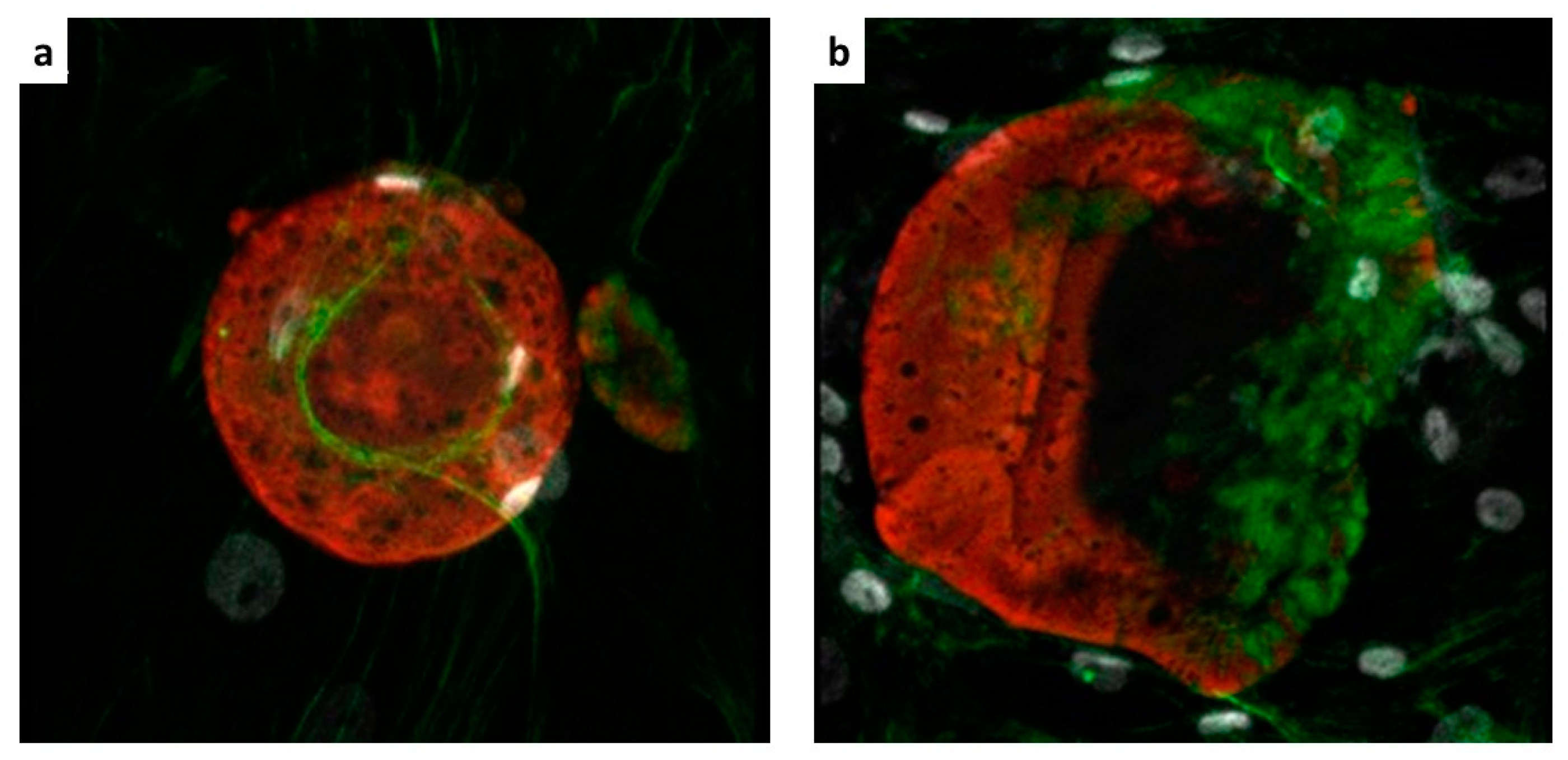
Confocal microscopy was performed for the detection of the hMSCs on the microparticles by means of a Zeiss LSM 5 Pascal Model Confocal Microscope (Carl Zeiss International, Oberkochen, Germany). Ten ILCSs were recovered and stained with phalloidin-FITC, incubated for 1 h at room temperature (Sigma-Aldrich), and with Hoechst 33,258 dye (Sigma-Aldrich) for nuclear staining. Subsequently, ILCSs were seeded on microscope slides.
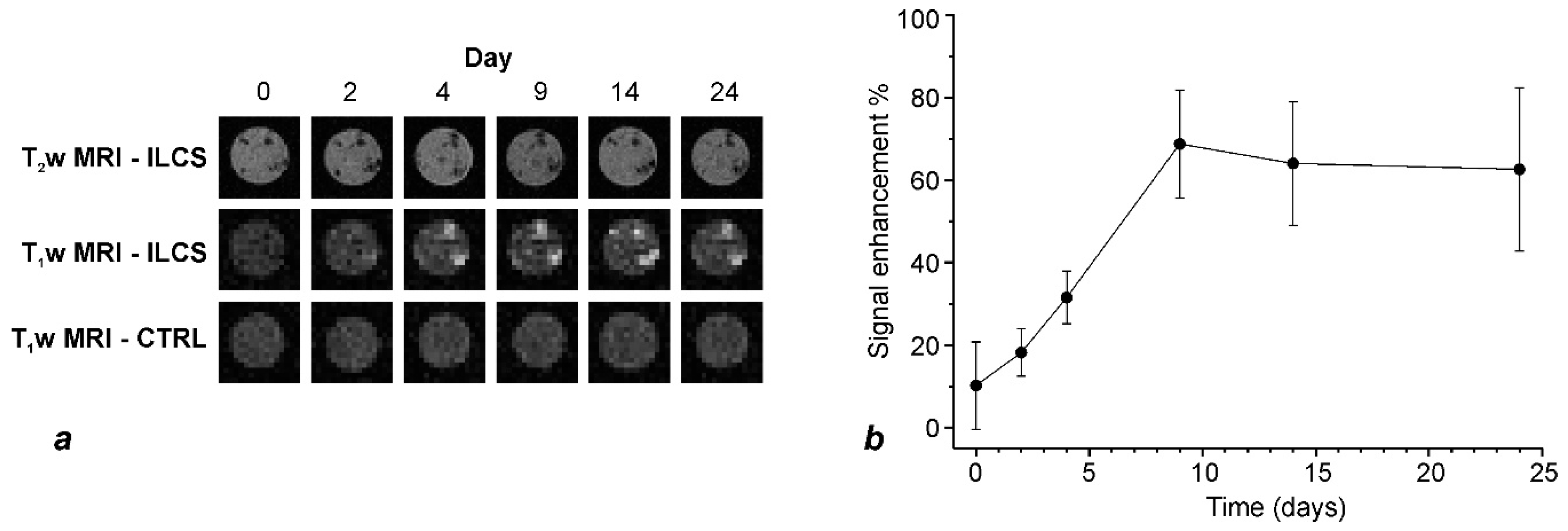
4.4. Release of Gd-NPs In Vitro
To evaluate the release of Gd-NPs in vitro, eight mg of microparticles were embedded into 1% agar contained in 0.5 mL Eppendorf tube. The pH was kept at 7.4 (HEPES buffer, Sigma, St. Louis, MO, USA). The volume of gelified agar was 150 μL. Twenty μL of HEPES buffer were placed on top of the agar to keep the agar well hydrated. MR images were acquired with a Bruker Avance microimaging MR scanner operating at 300 MHz (7 T). T1 weighted images were acquired with a mic_msme (multislice-multiecho) pulse sequence, with TR 200 ms, TE 3.6 ms, FOV = 3.5 × 3.5 cm, matrix 128 × 128, 15 slices 0.5 mm thick. T2 weighted images were acquired with a mic_RARE pulse sequence, with TR 4000 ms, TE 4.4 ms, FOV = 3.5 × 3.5 cm, matrix 256 × 256, 15 slices 0.5 mm thick. MRI images were acquired up to day 24.
4.5. Animal Studies
Animal studies were conducted in accordance with National Institute of Health Guidelines for the Care and Use of Laboratory Animals. All procedures were approved by the Ethics Committee of the University of Turin and the Italian Health Ministry (authorization number Prot. CC652.43). Mice were kept in our institutional animal facility under controlled conditions of temperature, humidity with access to food and water ad libitum.
4.5.1. Cell Implants
Implants to be grafted in mice as described in
Scheme 1 were prepared by seeding hMSCs on scaffolds, as described in
Section 4.2.1. The cell-loaded scaffolds (or control cell-free scaffolds) were recovered and mixed with 150 μL of MatrigelTM (Becton Dickinson, Franklin Lakes, NJ, USA) and the mixture kept ice cold. Such a mixture solution was surgically implanted subcutaneously on the back of either immunocompromised NSG mice (
n = 2), or immunocompetent FVB mice (
n = 2). In detail, mice were anesthetized with i.m. injection of 20 mg kg
−1 tiletamine–zolazepam (Zoletil 100; Virbac, Milan, Italy) mixed with 5 mg kg
−1 xylazine (Rompun; Bayer, Milan, Italy) and, under sterile conditions, a small skin incision was made on the back; the mixture was implanted using a large needle. Each mouse received two subcutaneous implants, one implant contained scaffolded hMSCs (+hMSC, placed on the left flank), and the other implant contained the cell-free scaffolds only (−hMSC, right flank). Follow-up by MRI started at Day 0, i.e., the day after surgery. The animals were followed up for 18 days by MRI and sacrificed at Day 20. The implants were excised and subjected to histology.
4.5.2. Magnetic Resonance Imaging
Prior to the acquisition of MR images, mice were anesthetized by i.m. administration of Zoletil and Rompun (see above). MR images were acquired with a Bruker Avance microimaging MR scanner (Bruker BioSpin, Rheinstetten, Germany) operating at 300 MHz (7 T). T1 weighted images were acquired with a mic_msme pulse sequence, with TR 200 ms, TE 4.7 ms, FOV = 3.5 × 3.5 cm, matrix 256 × 256, 13 slices 1 mm thick. T2 weighted images were acquired with a mic_RARE pulse sequence, with TR 2600 ms, TE 36 ms, FOV = 3.5 × 3.5 cm, matrix 384 × 384, 13 slices 1 mm thick. T1w and T2w MRI images were acquired at day 0, 5, 7, 10, 12 and 18 post implantation. Image reconstruction was performed with Paravision 5.1 and image analysis by ImageJ 1.47v.
T
2w MR images at each time point were used to identify the regions of interest (ROI) within matrigel that contained ILCSs. The Signal Enhancement (SE%) was then calculated on the corresponding ROI on co-registered T
1w MR images according to the formula:
where SI
ROI is the signal intensity measured in the ROI drawn around the ILCSs, and SI
ref is the signal intensity measured on a reference sample. The reference sample was a vial containing phosphate buffered saline, placed next to the imaged animal. As mice were imaged at different times, it was not always possible to take and compare slices exactly at the same position. To have a consistent quantitation of the time course of contrast enhancement, a set of 13 axial slices (each one mm thick) were placed such to have a complete coverage of both +hMSC and –hMSC implants (
Figure S8). T
2w and T
1w MR images were co-registered. T
2w images were used to define the ROI around ILCS, while T
1w weighted images were used to measure the gadolinium dependent SE due to released Gd-NPs. The change of SE was averaged on the entire volume of the implant at every time point in the serial images (with exclusion of slices suspected of partial volume artefacts; the smallest implant spanned across four valid slices at least).
4.5.3. Histology
Cell implants were excised from mice at the end of the follow-up period (Day 20), and explants were cut into 5 μm-thick sections. Sections were stained with hematoxylin and eosin (H&E) or Masson trichrome, or Sirius red. Fluorescence confocal microscopy was performed for the detection of the hMSCs in the implant. Sections were labelled with rabbit anti-HLA antibody (Cod, ab52922 Abcam, Cambridge Science Park, UK). Alexa Fluor anti-rabbit antibody (Molecular Probes, Leiden, the Netherlands) was used as secondary antibody. Hoechst dye was added for nuclear staining. Microscopy analysis was performed using an Aptome Zeiss (Carl Zeiss International, Oberkochen, Germany).



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}