Human Adipose Stem Cells (hASCs) Grown on Biodegradable Microcarriers in Serum- and Xeno-Free Medium Preserve Their Undifferentiated Status

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microcarriers Production
2.2. Extraction of Adipo-Cutaneous Tissue
2.3. Isolation and Culture of hASCs
2.3.1. Isolation of Stromal Vascular Fraction
2.3.2. Characterization of SVF Cells by Flow Cytometry
2.3.3. Flow Cytometer Procedure
2.3.4. Initial Seeding of the SVF Cells and Cell Passaging
2.4. Cells Seeding on MCs for Static Experiments
2.5. Attachment and Growth of hASCs in Static Conditions
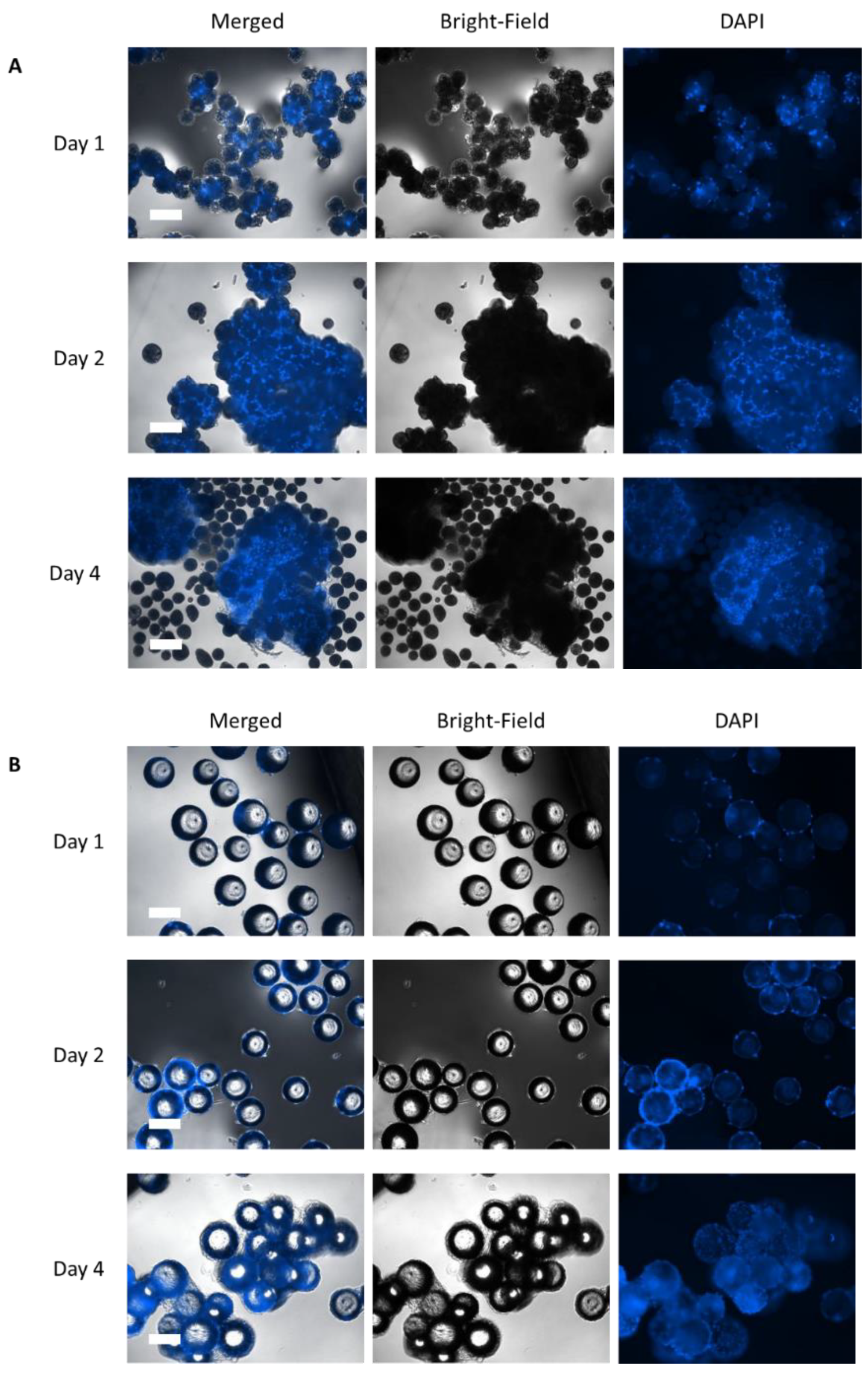
2.5.1. Cell Adhesion—Nuclei Analysis
2.5.2. Cells Adhesion—Scanning Electron Microscope (SEM) Analysis
2.5.3. Analysis of Nuclei Released from hASCs Grown on MCs by Flow Cytometry
2.6. Evaluation of Cellular Phenotype
2.6.1. Cell Detachment and Harvesting from the MCs for Flow Cytometry or RT-qPCR Analysis
2.6.2. Flow Cytometry Analysis of hASCs Grown on Cell Culture Vessels or MCs
2.6.3. RT-qPCR Analysis
2.7. Proof-of-Concept Cultivation in Spinner Flasks
2.8. Biochemical Analysis
2.8.1. Proteome Profiler Array
2.8.2. Amino Acid Analysis
2.9. Statistical Analysis
3. Results
3.1. Properties and Stability of Microcarriers
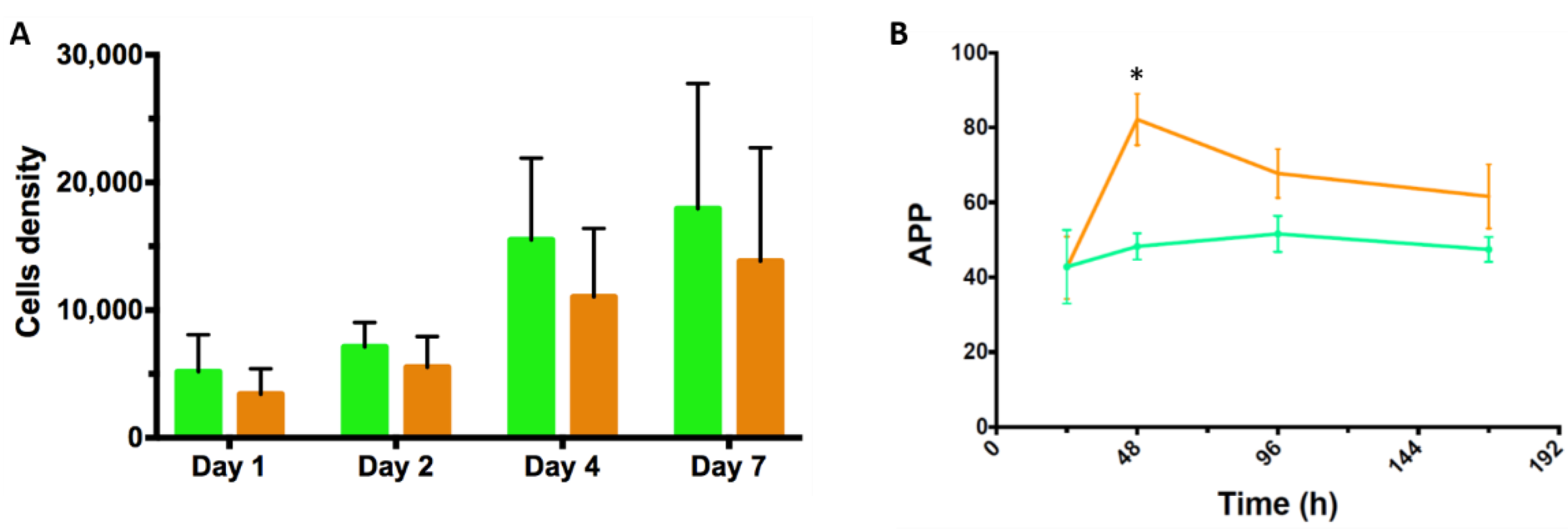
3.2. Static Investigations: Cell Adhesion and Proliferation on MCs
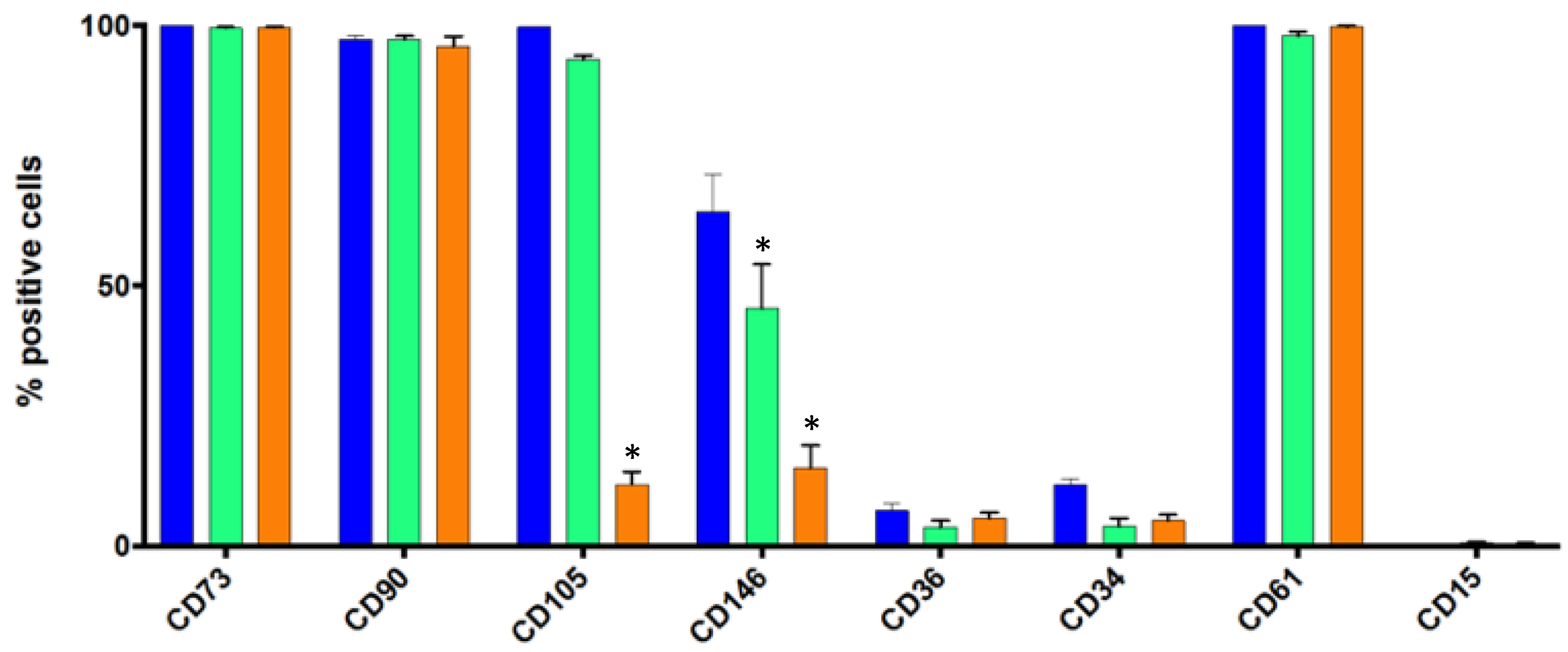
3.3. Flow Cytometry Analysis of Some Standard Markers in Static Conditions
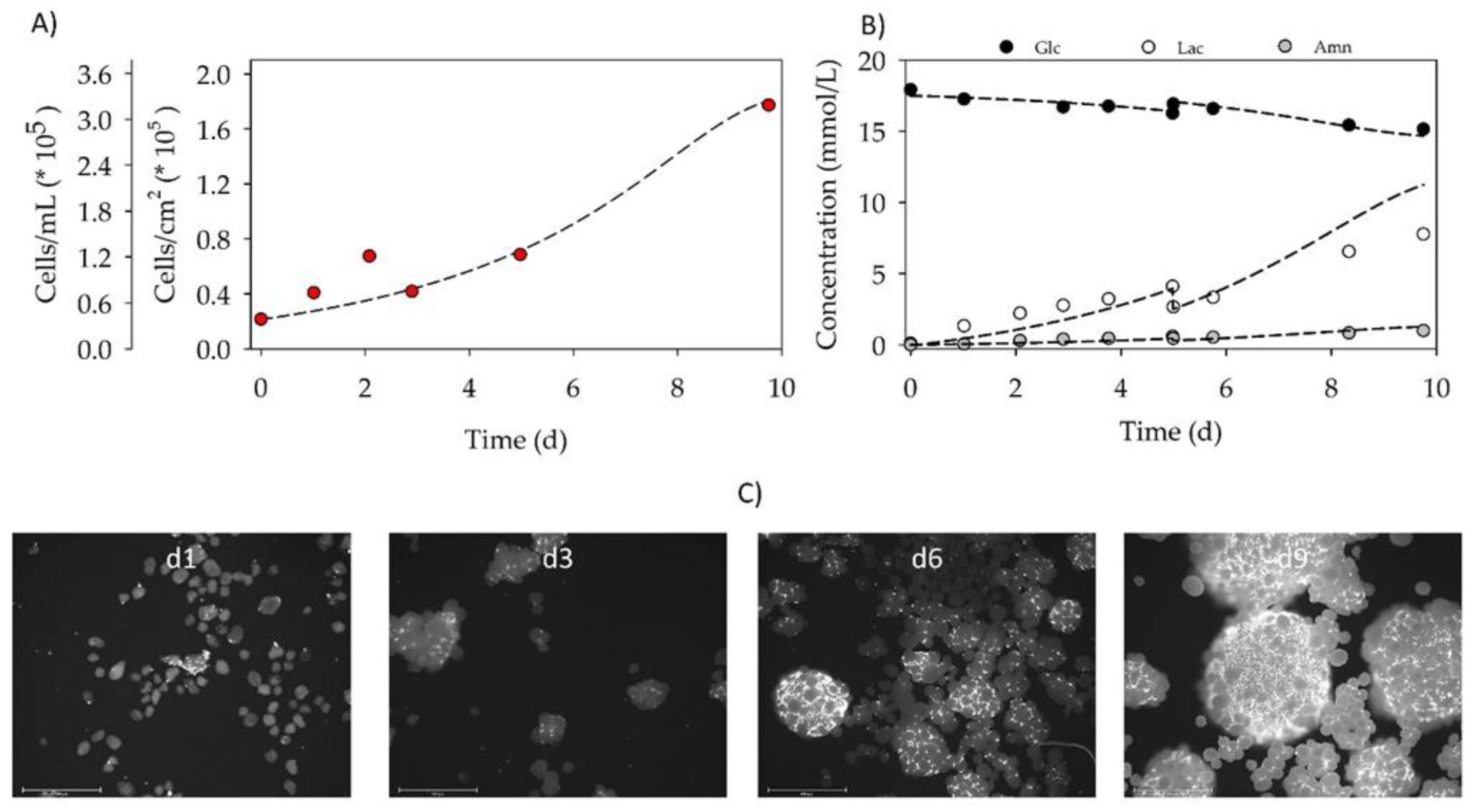
3.4. Proof-of-Concept Cultivation in Small Scale Spinner Flasks
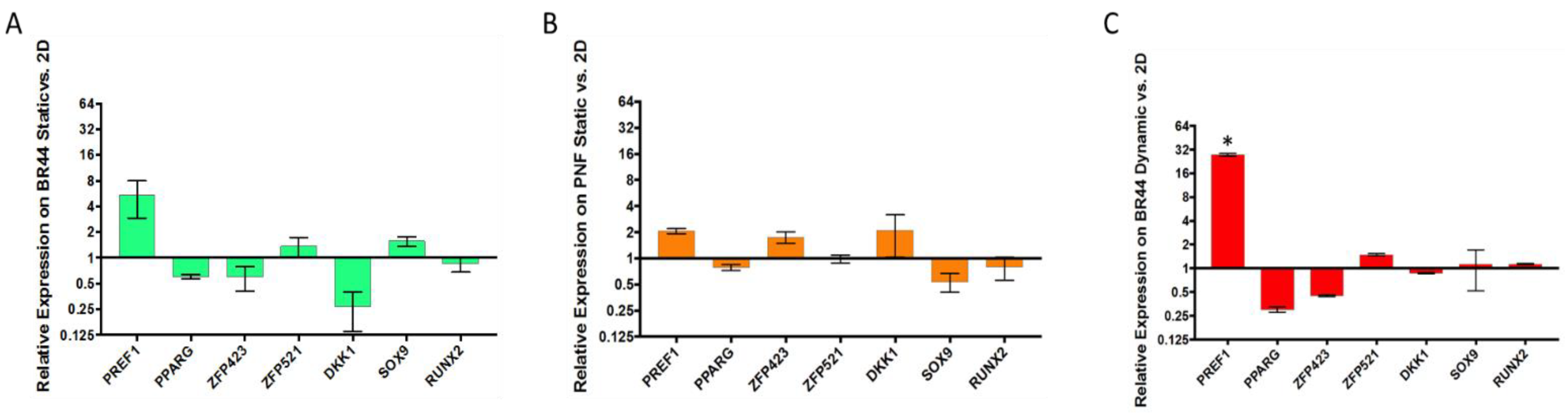
3.5. Gene Expression Survey by RT-qPCR of Some Essential Factors Regulating Adipogenesis
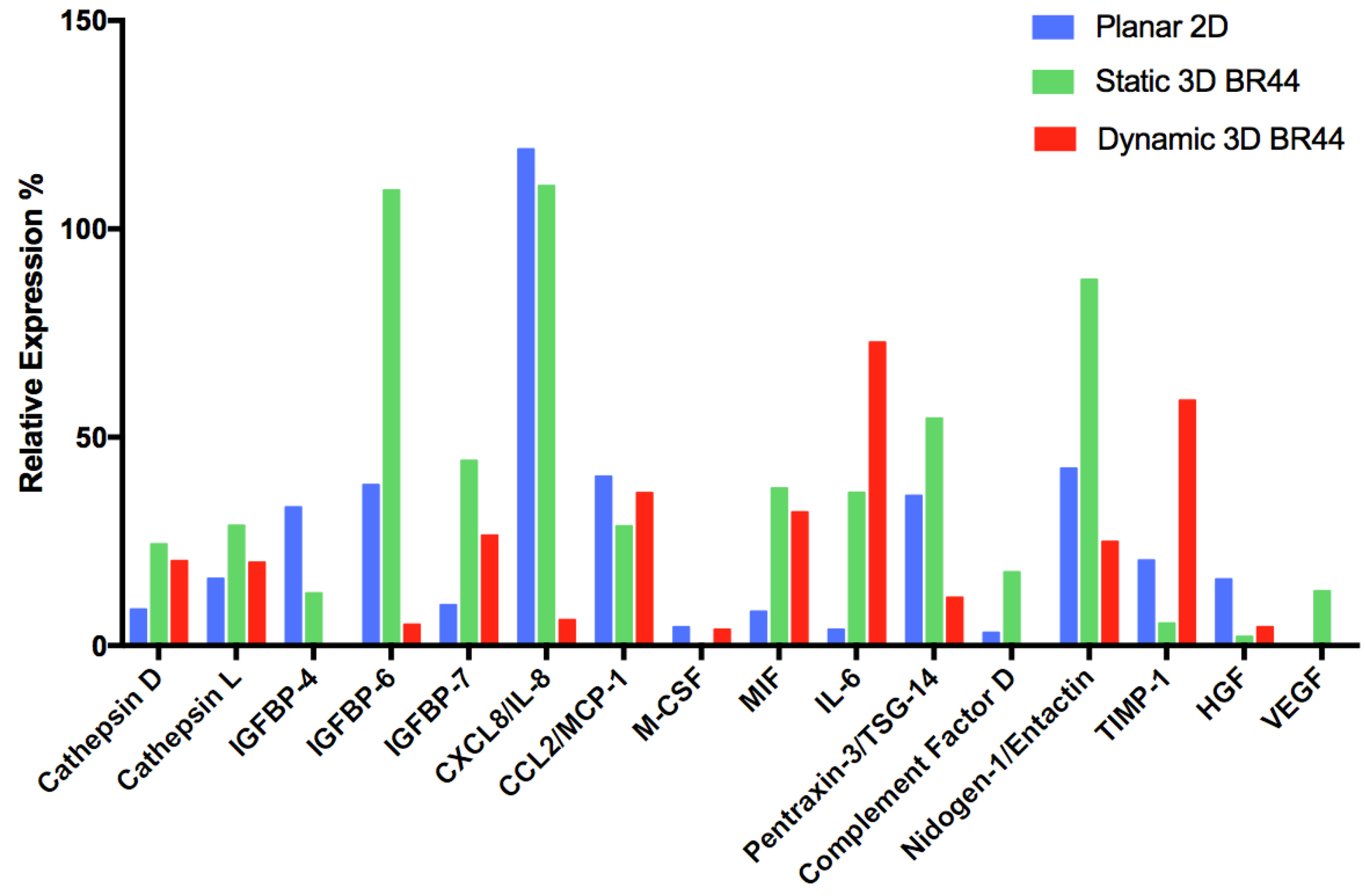
3.6. Investigating the Secretome Profile of hASCs Cultured in Standard Static 2D vs. Static or Dynamic 3D Conditions
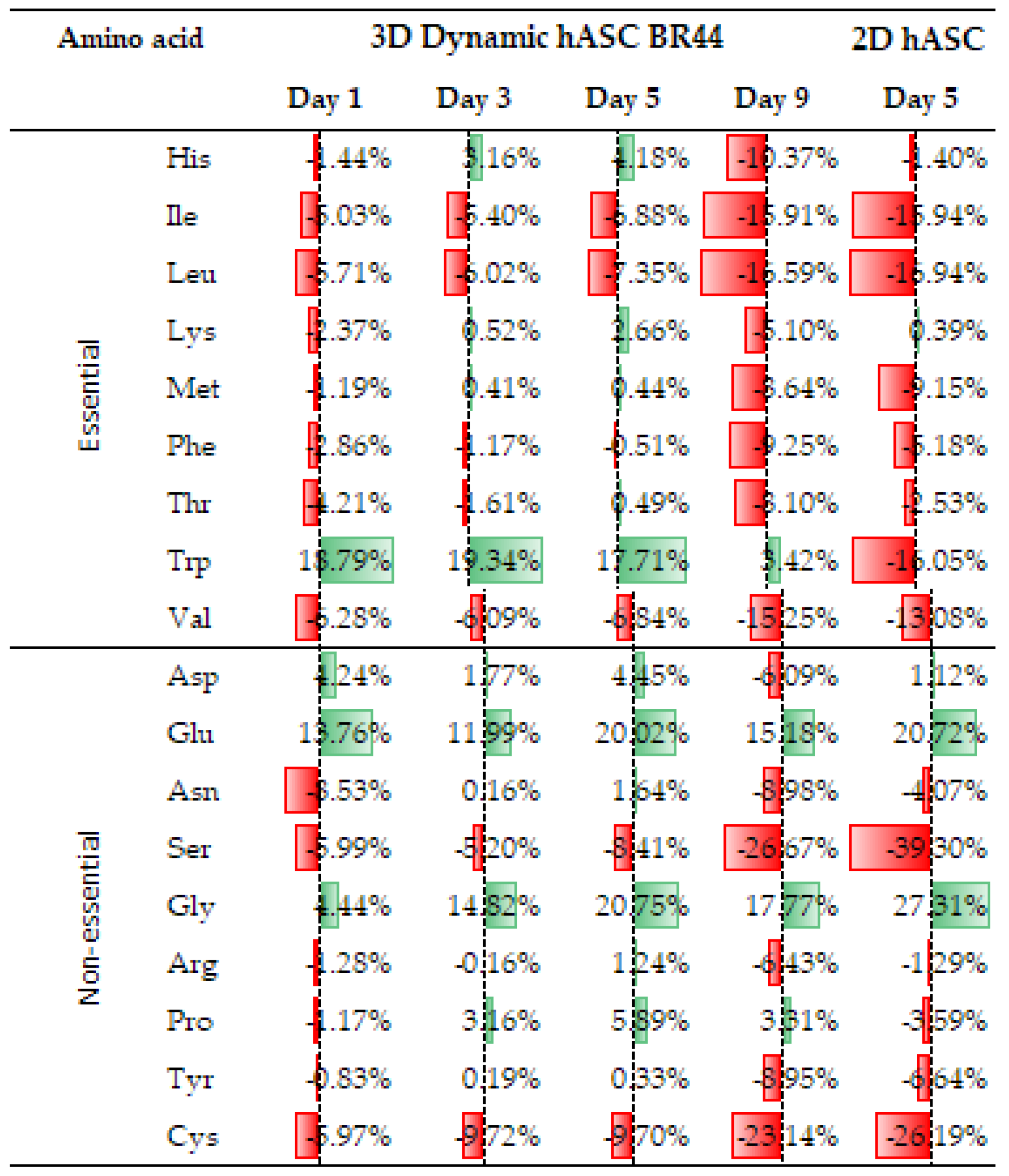
3.7. Comparing the Amino Acid Consumption between hASCs Cultured in a Standard 2D System with Those Grown in Dynamic 3D Conditions on BR44 MC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosen, E.D.; Spiegelman, B.M. What we talk about when we talk about fat. Cell 2014, 156, 20–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheele, C.; Wolfrum, C. Brown Adipose Crosstalk in Tissue Plasticity and Human Metabolism. Endocr. Rev. 2020, 41, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebo, Z.L.; Rodeheffer, M.S. Assembling the adipose organ: Adipocyte lineage segregation and adipogenesis in vivo. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Bourin, P.; Bunnell, B.A.; Casteilla, L.; Dominici, M.; Katz, A.J.; March, K.L.; Redl, H.; Rubin, J.P.; Yoshimura, K.; Gimble, J.M. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: A joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy 2013, 15, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Pikuła, M.; Marek-Trzonkowska, N.; Wardowska, A.; Renkielska, A.; Trzonkowski, P. Adipose tissue-derived stem cells in clinical applications. Expert Opin. Biol. Ther. 2013, 13, 1357–1370. [Google Scholar] [CrossRef]
- Nielsen, F.M.; Riis, S.E.; Andersen, J.I.; Lesage, R.; Fink, T.; Pennisi, C.P.; Zachar, V. Discrete adipose-derived stem cell subpopulations may display differential functionality after in vitro expansion despite convergence to a common phenotype distribution. Stem Cell Res. Ther. 2016, 7, 177. [Google Scholar] [CrossRef] [Green Version]
- Kokai, L.E.; Marra, K.; Rubin, J.P. Adipose stem cells: Biology and clinical applications for tissue repair and regeneration. Transl. Res. 2014, 163, 399–408. [Google Scholar] [CrossRef]
- Tan, K.Y.; Reuveny, S.; Oh, S.K.W. Recent advances in serum-free microcarrier expansion of mesenchymal stromal cells: Parameters to be optimized. Biochem. Biophys. Res. Commun. 2016, 473, 769–773. [Google Scholar] [CrossRef]
- Schop, D.; van Dijkhuizen-Radersma, R.; Borgart, E.; Janssen, F.W.; Rozemuller, H.; Prins, H.-J.; de Bruijn, J.D. Expansion of human mesenchymal stromal cells on microcarriers: Growth and metabolism. J. Tissue Eng. Regen. Med. 2010, 4, 131–140. [Google Scholar] [CrossRef]
- Hernández, Y.G.; Fischer, R.W. Serum-free culturing of mammalian cells—Adaption to and cryopreservation in fully defined media. ALTEX 2007, 24, 110–116. [Google Scholar] [CrossRef]
- Chase, L.G.; Lakshmipathy, U.; Solchaga, L.A.; Rao, M.S.; Vemuri, M.C. A novel serum-free medium for the expansion of human mesenchymal stem cells. Stem Cell Res. Ther. 2010, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Rauch, C. Alternatives to the use of fetal bovine serum: Human platelet lysates as a serum substitute in cell culture media. ALTEX 2011, 28, 305–316. [Google Scholar] [CrossRef]
- Riis, S.; Nielsen, F.M.; Pennisi, C.P.; Zachar, V.; Fink, T. Comparative Analysis of Media and Supplements on Initiation and Expansion of Adipose-Derived Stem Cells. Stem Cells Transl. Med. 2016, 5, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.-C.; Jeske, R.; Chen, X.; Yuan, X.; Li, Y. Influence of Microenvironment on Mesenchymal Stem Cell Therapeutic Potency: From Planar Culture to Microcarriers. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, C.J.; Lee, K.; Nienow, A.W.; Thomas, R.J.; Smith, M.; Thomas, C.R. Expansion of human mesenchymal stem cells on microcarriers. Biotechnol. Lett. 2011, 33, 2325–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Zhang, K.; Yang, Y.; Deng, D.; Lyu, C.; Xu, H.; Liu, W.; Du, Y. Dispersible and Dissolvable Porous Microcarrier Tablets Enable Efficient Large-Scale Human Mesenchymal Stem Cell Expansion. Tissue Eng. Part C Methods 2020, 26, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.Y.; Teo, K.L.; Lim, J.F.Y.; Chen, A.K.L.; Reuveny, S.; Oh, S.K. Serum-free media formulations are cell line–specific and require optimization for microcarrier culture. Cytotherapy 2015, 17, 1152–1165. [Google Scholar] [CrossRef]
- Panella, S.; Muoio, F.; Jossen, V.; Harder, Y.; Eibl-Schindler, R.; Tallone, T. Chemically Defined Xeno- and Serum-Free Cell Culture Medium to Grow Human Adipose Stem Cells. Cells 2021, 10, 466. [Google Scholar] [CrossRef]
- Carvalho, P.P.; Wu, X.; Yu, G.; Dias, I.R.; Gomes, M.E.; Reis, R.L.; Gimble, J.M. The effect of storage time on adipose-derived stem cell recovery from human lipoaspirates. Cells Tissues Organs 2011, 194, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Muoio, F.; Panella, S.; Lindner, M.; Jossen, V.; Harder, Y.; Moccetti, T.; Eibl, R.; Müller, M.; Tallone, T. Development of a Biodegradable Microcarrier for the Cultivation of Human Adipose Stem Cells (hASCs) with a Defined Xeno- and Serum-Free Medium. Appl. Sci. 2021, 11, 925. [Google Scholar] [CrossRef]
- Bidlingmeyer, B.A.; Cohen, S.A.; Tarvin, T.L. Rapid analysis of amino acids using pre-column derivatization. J. Chromatogr. B Biomed. Sci. Appl. 1984, 336, 93–104. [Google Scholar] [CrossRef]
- Jossen, V.; Muoio, F.; Panella, S.; Harder, Y.; Tallone, T.; Eibl, R. An Approach towards a GMP Compliant In-Vitro Expansion of Human Adipose Stem Cells for Autologous Therapies. Bioengineering 2020, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, G.G.; Atashroo, D.A.; Maan, Z.N.; Hu, M.S.; Zielins, E.R.; Tsai, J.M.; Duscher, D.; Paik, K.; Tevlin, R.; Marecic, O.; et al. High-Throughput Screening of Surface Marker Expression on Undifferentiated and Differentiated Human Adipose-Derived Stromal Cells. Tissue Eng. Part A 2015, 21, 2281–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camilleri, E.T.; Gustafson, M.P.; Dudakovic, A.; Riester, S.M.; Garces, C.G.; Paradise, C.R.; Takai, H.; Karperien, M.; Cool, S.; Sampen, H.J.I.; et al. Identification and validation of multiple cell surface markers of clinical-grade adipose-derived mesenchymal stromal cells as novel release criteria for good manufacturing practice-compliant production. Stem Cell Res. Ther. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festy, F.; Hoareau, L.; Bes-Houtmann, S.; Péquin, A.M.; Gonthier, M.P.; Munstun, A.; Hoarau, J.J.; Césari, M.; Roche, R. Surface protein expression between human adipose tissue-derived stromal cells and mature adipocytes. Histochem. Cell Biol. 2005, 124, 113–121. [Google Scholar] [CrossRef]
- Christiaens, V.; Van Hul, M.; Lijnen, H.R.; Scroyen, I. CD36 promotes adipocyte differentiation and adipogenesis. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Volat, F.; Sandhow, L.; Galitzky, J.; Nguyen, T.; Esteve, D.; Åström, G.; Mejhert, N.; Ledoux, S.; Thalamas, C.; et al. CD36 Is a Marker of Human Adipocyte Progenitors with Pronounced Adipogenic and Triglyceride Accumulation Potential. Stem Cells 2017, 35, 1799–1814. [Google Scholar] [CrossRef] [Green Version]
- Jossen, V.; Eibl, R.; Kraume, M.; Eibl, D. Growth Behavior of Human Adipose Tissue-Derived Stromal/Stem Cells at Small Scale: Numerical and Experimental Investigations. Bioengineering 2018, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Higuera, G.; Schop, D.; Janssen, F.; van Dijkhuizen-Radersma, R.; van Boxtel, T.; van Blitterswijk, C.A. Quantifying In Vitro Growth and Metabolism Kinetics of Human Mesenchymal Stem Cells Using a Mathematical Model. Tissue Eng. Part A 2009, 15, 2653–2663. [Google Scholar] [CrossRef]
- Schop, D.; Janssen, F.W.; van Rijn, L.D.S.; Fernandes, H.; Bloem, R.M.; de Bruijn, J.D.; van Dijkhuizen-Radersma, R. Growth, Metabolism, and Growth Inhibitors of Mesenchymal Stem Cells. Tissue Eng. Part A 2009, 15, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, W.; Ding, J. Effects of L-lactic acid and D,L-lactic acid on viability and osteogenic differentiation of mesenchymal stem cells. Chin. Sci. Bull. 2013, 58, 2404–2411. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhou, Y.; Tan, W.-S. Influence of lactic acid on the proliferation, metabolism, and differentiation of rabbit mesenchymal stem cells. Cell Biol. Toxicol. 2009, 25, 573–586. [Google Scholar] [CrossRef]
- Hudak, C.S.; Gulyaeva, O.; Wang, Y.; Park, S.M.; Lee, L.; Kang, C.; Sul, H.S. Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Rep. 2014, 8, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Akerblad, P.; Kiviranta, R.; Gupta, R.K.; Kajimura, S.; Griffin, M.J.; Min, J.; Baron, R.; Rosen, E.D. Regulation of Early Adipose Commitment by Zfp521. PLoS Biol. 2012, 10, e1001433. [Google Scholar] [CrossRef]
- Wang, Y.; Sul, H.S. Pref-1 Regulates Mesenchymal Cell Commitment and Differentiation through Sox9. Cell Metab. 2009, 9, 287–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Arany, Z.; Seale, P.; Mepani, R.J.; Ye, L.; Conroe, H.M.; Roby, Y.A.; Kulaga, H.; Reed, R.R.; Spiegelman, B.M. Transcriptional control of preadipocyte determination by Zfp423. Nature 2010, 464, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, B.; Smith, U. The WNT Inhibitor Dickkopf 1 and Bone Morphogenetic Protein 4 Rescue Adipogenesis in Hypertrophic Obesity in Humans. Diabetes 2012, 61, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Christodoulides, C.; Laudes, M.; Cawthorn, W.P.; Schinner, S.; Soos, M.; O’Rahilly, S.; Sethi, J.K.; Vidal-Puig, A. The Wnt antagonist Dickkopf-1 and its receptors are coordinately regulated during early human adipogenesis. J. Cell Sci. 2006, 119, 2613–2620. [Google Scholar] [CrossRef] [Green Version]
- Lee, M. Hormonal Regulation of Adipogenesis. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; Volume 7, pp. 1151–1195. [Google Scholar]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef]
- Kapur, S.K.; Katz, A.J. Review of the adipose derived stem cell secretome. Biochimie 2013, 95, 2222–2228. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Holdsworth, S.R.; Gan, P. Cytokines: Names and Numbers You Should Care About. Clin. J. Am. Soc. Nephrol. 2015, 10, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Jiang, N.; Li, Y.; Shu, T.; Wang, J. Cytokines and inflammation in adipogenesis: An updated review. Front. Med. 2019, 13, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Qi, P.; Abdullahi, A.; Stanojcic, M.; Chen, P.; Parousis, A.; Amini-Nik, S.; Jeschke, M.G. Burn Induces Browning of the Subcutaneous White Adipose Tissue in Mice and Humans. Cell Rep. 2015, 13, 1538–1544. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Qu, X.; Liu, F.; Wang, C. Pentraxin 3 as a Prognostic Biomarker in Patients with Systemic Inflammation or Infection. Mediat. Inflamm. 2014, 2014, 421429. [Google Scholar] [CrossRef] [Green Version]
- Pattrick, M.; Luckett, J.; Yue, L.; Stover, C. Dual role of complement in adipose tissue. Mol. Immunol. 2009, 46, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, N.J.; Kim, S.; Jang, B.H.; Chang, S.H.; Yun, U.J.; Park, K.M.; Waki, H.; Li, D.Y.; Tontonoz, P.; Park, K.W. Small molecule-induced complement factor D (Adipsin) promotes lipid accumulation and adipocyte differentiation. PLoS ONE 2016, 11, e0162228. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26, 188–202. [Google Scholar] [CrossRef] [Green Version]
- Doverskog, M.; Ljunggren, J.; Öhman, L.; Häggström, L. Physiology of cultured animal cells. J. Biotechnol. 1997, 59, 103–115. [Google Scholar] [CrossRef]
- Schop, D. Growth and Metabolism of Mesenchymal Stem Cells Cultivated on Microcarriers. Ph.D. Thesis, University of Twente, Enschede, The Netherlands, 2010. [Google Scholar]
- Higuera, G.A.; Schop, D.; Spitters, T.W.G.M.; Van Dijkhuizen-Radersma, R.; Bracke, M.; De Bruijn, J.D.; Martens, D.; Karperien, M.; Van Boxtel, A.; Van Blitterswijk, C.A. Patterns of amino acid metabolism by proliferating human mesenchymal stem cells. Tissue Eng. Part A 2012, 18, 654–664. [Google Scholar] [CrossRef]
- Guilak, F.; Cohen, D.M.; Estes, B.T.; Gimble, J.M.; Liedtke, W.; Chen, C.S. Control of Stem Cell Fate by Physical Interactions with the Extracellular Matrix. Cell Stem Cell 2009, 5, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Dai, J.; Zhang, X.A. Environmental physical cues determine the lineage specification of mesenchymal stem cells. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Patrikoski, M.; Mannerström, B.; Miettinen, S. Perspectives for clinical translation of adipose stromal/stem cells. Stem Cells Int. 2019, 2019, 5858247. [Google Scholar] [CrossRef] [Green Version]
- Gstraunthaler, G. Alternatives to the use of fetal bovine serum: Serum-free cell culture. ALTEX Altern. Anim. Exp. 2003, 20, 275–281. [Google Scholar]
- Van der Valk, J.; Bieback, K.; Buta, C.; Cochrane, B.; Dirks, W.G.; Fu, J.; Hickman, J.J.; Hohensee, C.; Kolar, R.; Liebsch, M.; et al. Fetal Bovine Serum (FBS): Past-Present-Future. ALTEX Altern. Anim. Exp. 2018, 35, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Maggs, D.G.; Jacob, R.; Rife, F.; Lange, R.; Leone, P.; During, M.J.; Tamborlane, W.V.; Sherwin, R.S. Interstitial fluid concentrations of glycerol, glucose, and amino acids in human quadricep muscle and adipose tissue. Evidence for significant lipolysis in skeletal muscle. J. Clin. Investig. 1995, 96, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Fogh-Andersen, N.; Altura, M.; Altura, T.; Siggaard-Andersen, O. Composition of Interstitial Fluid. Clin. Chem. 1995, 41, 1522–1525. [Google Scholar] [CrossRef]
- Jossen, V.; van den Bos, C.; Eibl, R.; Eibl, D. Manufacturing human mesenchymal stem cells at clinical scale: Process and regulatory challenges. Appl. Microbiol. Biotechnol. 2018, 102, 3981–3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egger, D.; Tripisciano, C.; Weber, V.; Dominici, M.; Kasper, C. Dynamic cultivation of mesenchymal stem cell aggregates. Bioengineering 2018, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, Y.; Syková, E.; Kubinová, Š. The therapeutic potential of three-dimensional multipotent mesenchymal stromal cell spheroids. Stem Cell Res. Ther. 2017, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Leber, J.; Barekzai, J.; Blumenstock, M.; Pospisil, B.; Salzig, D.; Czermak, P. Microcarrier choice and bead-to-bead transfer for human mesenchymal stem cells in serum-containing and chemically defined media. Process Biochem. 2017, 59, 255–265. [Google Scholar] [CrossRef]
- Heathman, T.R.J.J.; Glyn, V.A.M.; Picken, A.; Rafiq, Q.A.; Coopman, K.; Nienow, A.W.; Kara, B.; Hewitt, C.J. Expansion, harvest and cryopreservation of human mesenchymal stem cells in a serum-free microcarrier process. Biotechnol. Bioeng. 2015, 112, 1696–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, L.H.; Vu, N.B.; Van Pham, P. The subpopulation of CD105 negative mesenchymal stem cells show strong immunomodulation capacity compared to CD105 positive mesenchymal stem cells. Biomed. Res. Ther. 2019, 6, 3131–3140. [Google Scholar] [CrossRef]
- Leroyer, A.S.; Blin, M.G.; Bachelier, R.; Bardin, N.; Blot-Chabaud, M.; Dignat-George, F. CD146 (Cluster of Differentiation 146): An Adhesion Molecule Involved in Vessel Homeostasis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, X. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 2013, 330, 150–162. [Google Scholar] [CrossRef]
- Duan, H.; Xing, S.; Luo, Y.; Feng, L.; Gramaglia, I.; Zhang, Y.; Lu, D.; Zeng, Q.; Fan, K.; Feng, J.; et al. Targeting endothelial CD146 attenuates neuroinflammation by limiting lymphocyte extravasation to the CNS. Sci. Rep. 2013, 3, 1687. [Google Scholar] [CrossRef] [Green Version]
- Xing, S.; Luo, Y.; Liu, Z.; Bu, P.; Duan, H.; Liu, D.; Wang, P.; Yang, J.; Song, L.; Feng, J.; et al. Targeting endothelial CD146 attenuates colitis and prevents colitis-associated carcinogenesis. Am. J. Pathol. 2014, 184, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef]
- Larochelle, C.; Cayrol, R.; Kebir, H.; Alvarez, J.I.; Lécuyer, M.A.; Ifergan, I.; Viel, É.; Bourbonnière, L.; Beauseigle, D.; Terouz, S.; et al. Melanoma cell adhesion molecule identifies encephalitogenic T lymphocytes and promotes their recruitment to the central nervous system. Brain 2012, 135, 2906–2924. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Duan, H.; Qian, Y.; Feng, L.; Wu, Z.; Wang, F.; Feng, J.; Yang, D.; Qin, Z.; Yan, X. Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res. 2017, 27, 352–372. [Google Scholar] [CrossRef] [Green Version]
- Durandt, C.; Van Vollenstee, F.A.; Dessels, C.; Kallmeyer, K.; De Villiers, D.; Murdoch, C.; Potgieter, M.; Pepper, M.S. Novel flow cytometric approach for the detection of adipocyte subpopulations during adipogenesis. J. Lipid Res. 2016, 57, 729–742. [Google Scholar] [CrossRef] [Green Version]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.; et al. CD 36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018, 19, e45274. [Google Scholar] [CrossRef] [PubMed]
- Breimer, M.E.; Säljö, K.; Barone, A.; Teneberg, S. Glycosphingolipids of human embryonic stem cells. Glycoconj. J. 2017, 34, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, J.K.; Draper, J.S.; Baillie, H.S.; Fishel, S.; Thomson, J.A.; Moore, H.; Andrews, P.W. Preimplantation Human Embryos and Embryonic Stem Cells Show Comparable Expression of Stage-Specific Embryonic Antigens. Stem Cells 2002, 20, 329–337. [Google Scholar] [CrossRef]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise review: Evidence for CD34 as a common marker for diverse progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.; Kurtz, A.; Barutcu, N.; Bodo, J.; Thiel, A.; Dong, J. Concerted regulation of CD34 and CD105 accompanies mesenchymal stromal cell derivation from human adventitial stromal cell. Stem Cells Dev. 2013, 22, 815–827. [Google Scholar] [CrossRef]
- Raajendiran, A.; Ooi, G.; Bayliss, J.; O’Brien, P.E.; Schittenhelm, R.B.; Clark, A.K.; Taylor, R.A.; Rodeheffer, M.S.; Burton, P.R.; Watt, M.J. Identification of Metabolically Distinct Adipocyte Progenitor Cells in Human Adipose Tissues. Cell Rep. 2019, 27, 1528–1540. [Google Scholar] [CrossRef] [Green Version]
- Mariman, E.C.M.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell. Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef] [Green Version]
- Masson, O.; Prébois, C.; Derocq, D.; Meulle, A.; Dray, C.; Daviaud, D.; Quilliot, D.; Valet, P.; Muller, C.; Liaudet-Coopman, E. Cathepsin-D, a key protease in breast cancer, is up-regulated in obese mouse and human adipose tissue, and controls adipogenesis. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, Y.; Pan, J.; Sun, J.; Liu, J.; Libby, P.; Sukhova, G.K.; Doria, A.; Katunuma, N.; Peroni, O.D.; et al. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat. Cell Biol. 2007, 9, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D-Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, N.J.; Slater, T.A.; Matthews, C.J.; Wheatcroft, S.B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 2019, 19, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Blüher, S.; Kratzsch, J.; Kiess, W. Insulin-like growth factor I, growth hormone and insulin in white adipose tissue. Best Pr. Res. Clin. Endocrinol. Metab. 2005, 19, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, X.; Dang, H.; Liu, P.; Zhang, B.O.; Xu, F. Insulin-like growth factor 2 regulates the proliferation and differentiation of rat adipose-derived stromal cells via IGF-1R and IR. Cytotherapy 2019, 21, 619–630. [Google Scholar] [CrossRef]
- Bäck, K.; Brännmark, C.; Strålfors, P.; Arnqvist, H.J. Differential effects of IGF-I, IGF-II and insulin in human preadipocytes and adipocytes—Role of insulin and IGF-I receptors. Mol. Cell. Endocrinol. 2011, 339, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Gealekman, O.; Gurav, K.; Chouinard, M.; Straubhaar, J.; Thompson, M.; Malkani, S.; Hartigan, C.; Corvera, S. Control of Adipose Tissue Expandability in Response to High Fat Diet by the Insulin-like Growth Factor-binding Protein-4. J. Biol. Chem. 2014, 289, 18327–18338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headey, S.J.; Leeding, K.S.; Norton, R.S.; Bach, L.A. Contributions of the N- and C-terminal domains of IGF binding protein-6 to IGF binding. J. Mol. Endocrinol. 2004, 33, 377–386. [Google Scholar] [CrossRef]
- Van Breevoort, D.; Van Agtmaal, E.L.; Dragt, B.S.; Gebbinck, J.K.; Dienava-Verdoold, I.; Kragt, A.; Bierings, R.; Horrevoets, A.J.G.; Valentijn, K.M.; Eikenboom, J.C.; et al. Proteomic screen identifies IGFBP7 as a novel component of endothelial cell-specific weibel-palade bodies. J. Proteome Res. 2012, 11, 2925–2936. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Murai, T.; Tanaka, T.; Yamaguchi, K.; Nagakubo, D.; Lee, C.M.; Kiyomi, M.; Tamura, S.; Matsuzawa, Y.; Miyasaka, M. Characterization of mac25/angiomodulin expression by high enothelial venule cells in lymphoid tissues and its identification as an inducible marker for activated endothelial cells. Int. Immunol. 2002, 14, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Nagakubo, D.; Murai, T.; Tanaka, T.; Usui, T.; Matsumoto, M.; Sekiguchi, K.; Miyasaka, M. A High Endothelial Venule Secretory Protein, Mac25/Angiomodulin, Interacts with Multiple High Endothelial Venule-Associated Molecules Including Chemokines. J. Immunol. 2003, 171, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Hooper, A.T.; Shmelkov, S.V.; Gupta, S.; Milde, T.; Bambino, K.; Gillen, K.; Goetz, M.; Chavala, S.; Baljevic, M.; Murphy, A.J.; et al. Angiomodulin Is a Specific Marker of Vasculature and Regulates Vascular Endothelial Growth Factor-A-Dependent Neoangiogenesis. Circ. Res. 2009, 105, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef]
- Atsumi, T.; Cho, Y.; Leng, L.; McDonald, C.; Yu, T.; Danton, C.; Hong, E.; Mitchell, R.A.; Metz, C.; Niwa, H.; et al. The Proinflammatory Cytokine Macrophage Migration Inhibitory Factor Regulates Glucose Metabolism during Systemic Inflammation. J. Immunol. 2007, 179, 5399–5406. [Google Scholar] [CrossRef] [Green Version]
- Calandra, T.; Bernhagen, J.; Metz, C.N.; Spiegel, L.A.; Bacher, M.; Donnelly, T.; Cerami, A.; Bucala, R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature 1995, 377, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Meissburger, B.; Stachorski, L.; Röder, E.; Rudofsky, G.; Wolfrum, C. Tissue inhibitor of matrix metalloproteinase 1 (TIMP1) controls adipogenesis in obesity in mice and in humans. Diabetologia 2011, 54, 1468–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, C.G.; Jia, Y.L.; Hu, L. Tissue Inhibitor of Metalloprotease-1 (TIMP-1) Regulates Adipogenesis of Adipose-derived Stem Cells (ASCs) via the Wnt Signaling Pathway in an MMP-independent Manner. Curr. Med. Sci. 2020, 40, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Ries, C. Cytokine functions of TIMP-1. Cell. Mol. Life Sci. 2014, 71, 659–672. [Google Scholar] [CrossRef]
- Grünwald, B.; Schoeps, B.; Krüger, A. Recognizing the Molecular Multifunctionality and Interactome of TIMP-1. Trends Cell Biol. 2019, 29, 6–19. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef]
- Meijer, A.J. Amino acids as regulators and components of nonproteinogenic pathways. J. Nutr. 2003, 133, 2057–2062. [Google Scholar] [CrossRef]
- Nesterov, S.V.; Yaguzhinsky, L.S.; Podoprigora, G.I.; Nartsissov, Y.R. Amino Acids as Regulators of Cell Metabolism. Biochemistry 2020, 85, 393–408. [Google Scholar] [CrossRef]
- Menta, R.; Mancheño-Corvo, P.; Del Río, B.; Ramírez, C.; DelaRosa, O.; Dalemans, W.; Lombardo, E. Tryptophan concentration is the main mediator of the capacity of adipose mesenchymal stromal cells to inhibit T-lymphocyte proliferation in vitro. Cytotherapy 2014, 16, 1679–1691. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.; Murphy, J.M.; Mahon, B.P. Interferon-γ does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin. Exp. Immunol. 2007, 149, 353–363. [Google Scholar] [CrossRef]
- Yao, T.; Asayama, Y. Animal-cell culture media: History, characteristics, and current issues. Reprod. Med. Biol. 2017, 16, 99–117. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | μ | td | Atteff | Xmax | EF | YLac/Glc | qGlc | qLac | qAmn |
|---|---|---|---|---|---|---|---|---|---|
| (d−1) | (d) | (%) | (105 cells/mL) | (-) | (mmol/mmol) | (pmol/cell/d) | (pmol/cell/d) | (pmol/cell/d) | |
| BR44 | 0.25 | 2.8 | 188 | 3.18 | 8.1 | 2.6 | 3.60 | 10.01 | 1.87 |
| Secreted Protein Type | Analyte/Control | 2D hASC | 3D Static hASC BR44 | 3D Dynamic hASC BR44 |
|---|---|---|---|---|
| Aspartyl- and cysteine-proteases | Cathepsin D | X | XX | XX |
| Cathepsin L | X | XX | X | |
| Insulin-like growth factor binding proteins | IGFBP-4 | XX | X | (-) |
| IGFBP-6 | X | XXX | (X) | |
| IGFBP-7 | X | XXX | XX | |
| Cytokines, chemokines, inflammatory factors | CXCL8/IL-8 | XXX | XXX | (X) |
| CCL2/MCP-1 | X | X | X | |
| M-CSF | X | (-) | X | |
| MIF | (X) | XXX | XXX | |
| IL-6 | (X) | XX | XXX | |
| Pentraxin-3/TSG-14 | XX | XXX | X | |
| Complement Factor D | X | XXX | (-) | |
| Extracellular matrix | Nidogen-1/Entactin | XX | XXX | X |
| Metalloproteases inhibitor | TIMP-1 | XX | X | XXX |
| Angiogenesis and cell proliferation | HGF | XX | (X) | X |
| VEGF | (-) | X | (-) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muoio, F.; Panella, S.; Jossen, V.; Lindner, M.; Harder, Y.; Müller, M.; Eibl, R.; Tallone, T. Human Adipose Stem Cells (hASCs) Grown on Biodegradable Microcarriers in Serum- and Xeno-Free Medium Preserve Their Undifferentiated Status. J. Funct. Biomater. 2021, 12, 25. https://doi.org/10.3390/jfb12020025
Muoio F, Panella S, Jossen V, Lindner M, Harder Y, Müller M, Eibl R, Tallone T. Human Adipose Stem Cells (hASCs) Grown on Biodegradable Microcarriers in Serum- and Xeno-Free Medium Preserve Their Undifferentiated Status. Journal of Functional Biomaterials. 2021; 12(2):25. https://doi.org/10.3390/jfb12020025
Chicago/Turabian StyleMuoio, Francesco, Stefano Panella, Valentin Jossen, Matias Lindner, Yves Harder, Michele Müller, Regine Eibl, and Tiziano Tallone. 2021. "Human Adipose Stem Cells (hASCs) Grown on Biodegradable Microcarriers in Serum- and Xeno-Free Medium Preserve Their Undifferentiated Status" Journal of Functional Biomaterials 12, no. 2: 25. https://doi.org/10.3390/jfb12020025