
Polyamide/Poly(Amino Acid) Polymers for Drug Delivery

 , , ,
, , ,  and
and
Abstract
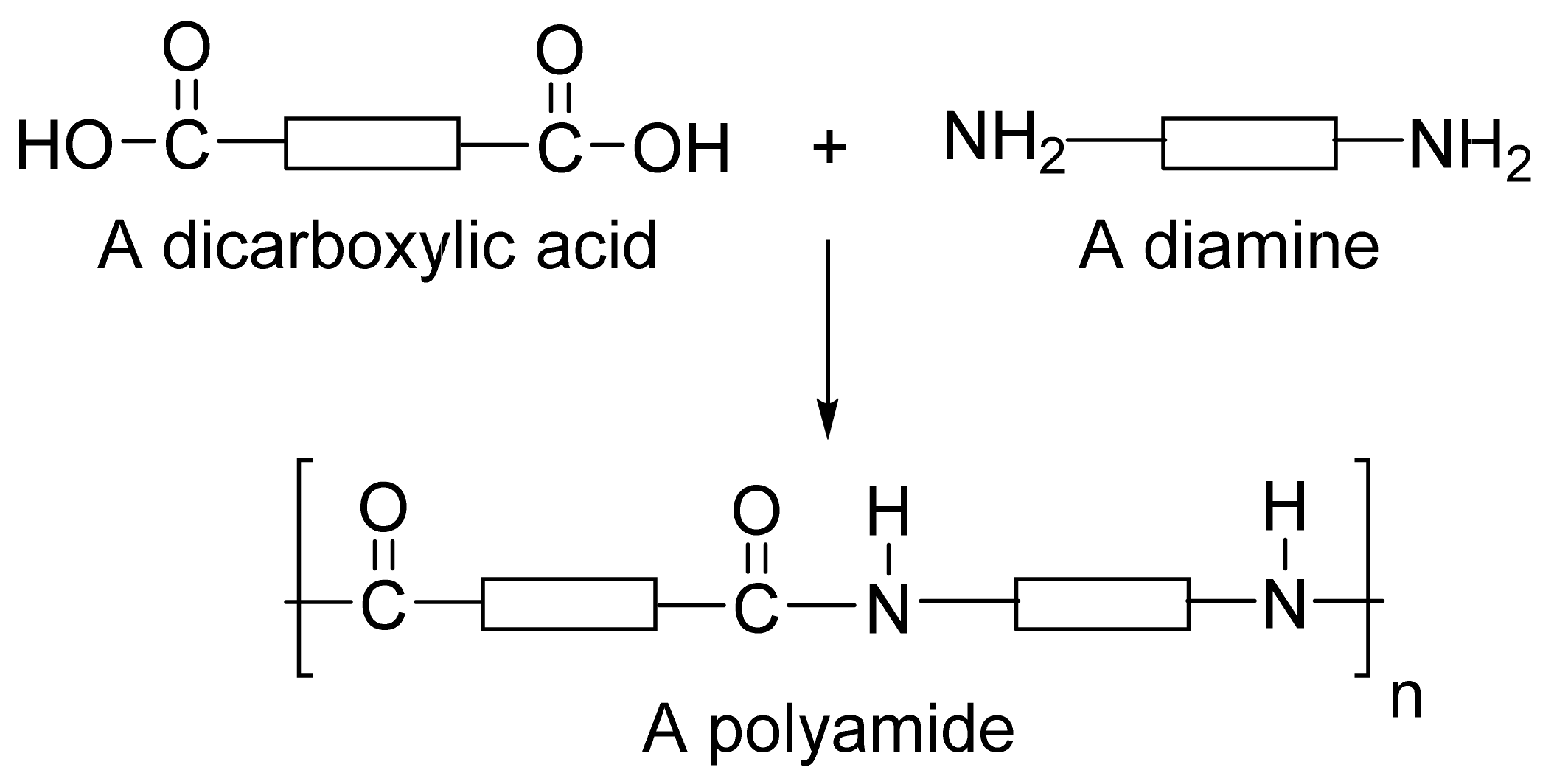
:1. Introduction
2. Polyamide Polymers Used in Drug Delivery
2.1. Poly(Aspartic Acid)
2.2. Poly-L-Lysine
2.3. Poly(Glutamic Acid)
2.4. Poly(Amidoamine)
2.5. Fatty Acid-Based Polyamide
2.6. Others
3. Poly(Amino Acid)-Based Micelles Undergoing Clinical Trials
3.1. NK 105, Paclitaxel Incorporating Micelles
3.1.1. Method of Preparation
3.1.2. Pharmacokinetics and Pharmacodynamics of NK105
3.1.3. In Vivo Pharmacodynamic and Toxicity Study of NK105
3.1.4. Clinical Studies
3.1.5. Other Similar Products in the Pipeline
3.2. NC-6004, Cisplatin-Incorporating Micellar Nanoparticle
3.2.1. Preparation of NC-6004 Micellar Complex
3.2.2. Pharmacokinetics and Pharmacodynamics
3.2.3. In Vivo Anti-Tumor Activity
3.2.4. Nephrotoxicity of Cisplatin and NC-6004
3.2.5. Clinical Studies of NC-6004
3.2.6. Other Similar Products in the Pipeline
4. Challenges and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for Drug Delivery Systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gachard, I.; Bechaouch, S.; Coutin, B.; Sekiguchi, H. Drug delivery from nonpeptidic α-amino acid containing polyamides. Polym. Bull. 1997, 38, 427–431. [Google Scholar] [CrossRef]
- Song, R.; Murphy, M.; Li, C.; Ting, K.; Soo, C.; Zheng, Z. Current development of biodegradable polymeric materials for biomedical applications. Drug Des. Dev. Ther. 2018, 12, 3117–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabard, M.; Gouanvé, F.; Espuche, E.; Fulchiron, R.; Fillot, L.-A.; Trouillet-Fonti, L. Erasure of the processing effects in polyamide 6 based cast films by the introduction of montmorillonite: Effect on water and oxygen transport properties. J. Membr. Sci. 2014, 456, 11–20. [Google Scholar] [CrossRef]
- Winnacker, M. Polyamides and their functionalization: Recent concepts for their applications as biomaterials. Biomater. Sci. 2017, 5, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Bankar, S.B.; Nimbalkar, P.R.; Chavan, P.V.; Singhal, R.S. Chapter 12—Microbial Polyamino Acids: An Overview for Commercial Attention. In Role of Materials Science in Food Bioengineering; Grumezescu, A.M., Holban, A.M., Eds.; Academic Press: London, UK, 2018; pp. 381–412. [Google Scholar] [CrossRef]
- Francisco, D.L.; Paiva, L.B.; Aldeia, W. Advances in polyamide nanocomposites: A review. Polym. Compos. 2018, 40, 851–870. [Google Scholar] [CrossRef]
- Khattab, S.N.; Naim, S.E.A.; El-Sayed, M.; El Bardan, A.A.; Elzoghby, A.; Bekhit, A.; El-Faham, A. Design and synthesis of new s-triazine polymers and their application as nanoparticulate drug delivery systems. New J. Chem. 2016, 40, 9565–9578. [Google Scholar] [CrossRef]
- Jana, P.; Shyam, M.; Singh, S.; Jayaprakash, V.; Dev, A. Biodegradable polymers in drug delivery and oral vaccination. Eur. Polym. J. 2020, 142, 110155. [Google Scholar] [CrossRef]
- González-Aramundiz, J.; Lozano, M.V.; Sousa-Hervés, A.; Fernandez-Megia, E.; Csaba, N. Polypeptides and polyaminoacids in drug delivery. Expert Opin. Drug Deliv. 2012, 9, 183–201. [Google Scholar] [CrossRef]
- Ho, D.K.; Nguyen, D.T.; Thambi, T.; Lee, D.S.; Huynh, D.P. Polyamide-based pH and temperature-responsive hydrogels: Synthesis and physicochemical characterization. J. Polym. Res. 2018, 26, 7. [Google Scholar] [CrossRef]
- Patravale, V.; Dandekar, P.; Jain, R. Clinical trials industrial aspects. In Nanoparticulate Drug Delivery; Patravale, V., Dandekar, P., Jain, R., Eds.; Woodhead Publishing: Cambridge, UK, 2012; pp. 191–207. [Google Scholar] [CrossRef]
- Simmons, P.; McElroy, T.; Allen, A.R. A Bibliometric Review of Artificial Extracellular Matrices Based on Tissue Engineering Technology Literature: 1990 through 2019. Materials 2020, 13, 2891. [Google Scholar] [CrossRef] [PubMed]
- Nair, L.S.; Laurencin, C.T. Polymers as Biomaterials for Tissue Engineering and Controlled Drug Delivery. Adv. Biochem. Eng. Biotechnol. 2005, 102, 47–90. [Google Scholar] [CrossRef]
- James, K.; Levene, H.; Parsons, J.R.; Kohn, J. Small changes in polymer chemistry have a large effect on the bone–implant interface: Evaluation of a series of degradable tyrosine-derived polycarbonates in bone defects. Biomaterials 1999, 20, 2203–2212. [Google Scholar] [CrossRef]
- Kim, J.; Magno, M.H.R.; Alvarez, P.; Darr, A.; Kohn, J.; Hollinger, J.O. Osteogenic Differentiation of Pre-Osteoblasts on Biomimetic Tyrosine-Derived Polycarbonate Scaffolds. Biomacromolecules 2011, 12, 3520–3527. [Google Scholar] [CrossRef]
- Numata, K. Poly(amino acid)s/polypeptides as potential functional and structural materials. Polym. J. 2015, 47, 537–545. [Google Scholar] [CrossRef]
- Choi, A.H.; Ben-Nissan, B. Sol-gel production of bioactive nanocoatings for medical applications. Part II: Current research and development. Nanomedicine 2007, 2, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.-Y.; Wang, Z.-Y.; Chen, C.; Mo, F.-K.; Xu, Y.-L.; Tang, L.-X.; Liang, J.-J. Poly(aspartic-acid) derivatives as polymeric micelle drug delivery systems. J. Appl. Polym. Sci. 2006, 101, 2871–2878. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, X.; Yuan, L.; Hu, J. PEG-g-poly(aspartamide-co-N,N-dimethylethylenediamino aspartamide): Synthesis, characterization and its application as a drug delivery system. Prog. Nat. Sci. 2009, 19, 1305–1310. [Google Scholar] [CrossRef]
- Zheng, Y.; Yang, W.; Wang, C.; Hu, J.; Fu, S.; Dong, L.; Wu, L.; Shen, X. Nanoparticles based on the complex of chitosan and polyaspartic acid sodium salt: Preparation, characterization and the use for 5-fluorouracil delivery. Eur. J. Pharm. Biopharm. 2007, 67, 621–631. [Google Scholar] [CrossRef]
- Xiong, X.-B.; Mahmud, A.; Uludaǧ, H.; Lavasanifar, A. Conjugation of Arginine-Glycine-Aspartic Acid Peptides to Poly(ethylene oxide)-b-poly(ε-caprolactone) Micelles for Enhanced Intracellular Drug Delivery to Metastatic Tumor Cells. Biomacromolecules 2007, 8, 874–884. [Google Scholar] [CrossRef]
- Hayashi, T.; Iwatsuki, M. Biodegradation of copoly(L-aspartic acid/L-glutamic acid) in vitro. Biopolymers 1990, 29, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.M.; Varaprasad, K.; Rao, K.M.; Babu, A.S.; Srinivasulu, M.; Naidu, S.V. A Novel Biodegradable Green Poly(l-Aspartic Acid-Citric Acid) Copolymer for Antimicrobial Applications. J. Polym. Environ. 2011, 20, 17–22. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, G.; Fan, Y.; Ma, J. pH-Responsive Self-Assembly and conformational transition of partially propyl-esterified poly(α,β-l-aspartic acid) as amphiphilic biodegradable polyanion. Colloids Surf. B Biointerfaces 2009, 68, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Roweton, S.; Huang, S.J.; Swift, G. Poly(aspartic acid): Synthesis, biodegradation, and current applications. J. Environ. Polym. Degrad. 1997, 5, 175–181. [Google Scholar] [CrossRef]
- Ponta, A.; Bae, Y. PEG-poly(amino acid) Block Copolymer Micelles for Tunable Drug Release. Pharm. Res. 2010, 27, 2330–2342. [Google Scholar] [CrossRef]
- Tummala, S.; Kumar, M.S.; Prakash, A. Formulation and characterization of 5-Fluorouracil enteric coated nanoparticles for sustained and localized release in treating colorectal cancer. Saudi Pharm. J. 2014, 23, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Hou, T.; Li, J.; Yu, S.; Xu, Z.; Yin, M.; Wang, J.; Wang, X. Self-Propelled and Targeted Drug Delivery of Poly(aspartic acid)/Iron–Zinc Microrocket in the Stomach. ACS Nano 2019, 13, 1324–1332. [Google Scholar] [CrossRef]
- Thombre, S.M.; Sarwade, B.D. Synthesis and Biodegradability of Polyaspartic Acid: A Critical Review. J. Macromol. Sci. Part A 2005, 42, 1299–1315. [Google Scholar] [CrossRef]
- Adelnia, H.; Blakey, I.; Little, P.J.; Ta, H.T. Hydrogels Based on Poly(aspartic acid): Synthesis and Applications. Front. Chem. 2019, 7, 755. [Google Scholar] [CrossRef]
- Shima, S.; Matsuoka, H.; Iwamoto, T.; Sakai, H. Antimicrobial action of EPSILON-poly-L-lysine. J. Antibiot. 1984, 37, 1449–1455. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Wu, C. Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. J. Biol. Chem. 1987, 262, 4429–4432. [Google Scholar] [CrossRef]
- Mislick, K.A.; Baldeschwieler, J.D.; Kayyem, J.F.; Meade, T.J. Transfection of Folate-Polylysine DNA Complexes: Evidence for Lysosomal Delivery. Bioconj. Chem. 1995, 6, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M.; Langle-Rouault, F.; Kirlappos, H.; Wagner, E.; Mechtler, K.; Zenke, M.; Beug, H.; Birnstiel, M.L. Transferrin-polycation-mediated introduction of DNA into human leukemic cells: Stimulation by agents that affect the survival of transfected DNA or modulate transferrin receptor levels. Proc. Natl. Acad. Sci. USA 1990, 87, 4033–4037. [Google Scholar] [CrossRef] [Green Version]
- Perales, J.C.; Ferkol, T.; Beegen, H.; Ratnoff, O.D.; Hanson, R.W. Gene transfer in vivo: Sustained expression and regulation of genes introduced into the liver by receptor-targeted uptake. Proc. Natl. Acad. Sci. USA 1994, 91, 4086–4090. [Google Scholar] [CrossRef] [Green Version]
- Pillai, O.; Panchagnula, R. Polymers in drug delivery. Curr. Opin. Chem. Biol. 2001, 5, 447–451. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.H.; Joe, C.O.; Park, T.G. Folate receptor mediated intracellular protein delivery using PLL–PEG–FOL conjugate. J. Control. Release 2005, 103, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.H.; Chun, K.W.; Park, T.G. Target-Specific Cellular Uptake of PLGA Nanoparticles Coated with Poly(l-lysine)−Poly(ethylene glycol)−Folate Conjugate. Langmuir 2005, 21, 8852–8857. [Google Scholar] [CrossRef]
- Yang, D.H.; Kim, H.J.; Park, K.; Kim, J.K.; Chun, H.J. Preparation of poly-l-lysine-based nanoparticles with pH-sensitive release of curcumin for targeted imaging and therapy of liver cancer in vitro and in vivo. Drug Deliv. 2018, 25, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Fan, Y.; Li, Y.; Huang, L. Strategies on the nuclear-targeted delivery of genes. J. Drug Target. 2013, 21, 926–939. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Gao, W.; Gu, Y.; Zhang, W.; Cao, M.; Song, C.; Zhang, P.; Sun, M.; Yang, C.; Wang, S. Functions of poly-gamma-glutamic acid (γ-PGA) degradation genes in γ-PGA synthesis and cell morphology maintenance. Appl. Microbiol. Biotechnol. 2014, 98, 6397–6407. [Google Scholar] [CrossRef] [PubMed]
- Li, C. Poly(l-glutamic acid)–anticancer drug conjugates. Adv. Drug Deliv. Rev. 2002, 54, 695–713. [Google Scholar] [CrossRef]
- Richard, A.; Margaritis, A. Poly(glutamic Acid) for Biomedical Applications. Crit. Rev. Biotechnol. 2001, 21, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br. J. Cancer 2005, 93, 678–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, N.; Okazaki, S.; Cabral, H.; Miyamoto, M.; Kato, Y.; Sugiyama, Y.; Nishio, K.; Matsumura, Y.; Kataoka, K. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003, 63, 8977–8983. [Google Scholar] [PubMed]
- Stirland, D.L.; Nichols, J.W.; Miura, S.; Bae, Y.H. Mind the gap: A survey of how cancer drug carriers are susceptible to the gap between research and practice. J. Control. Release 2013, 172, 1045–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk-Wolthuis, W.N.E.; Tsang, S.K.Y.; Kettenes-Van Den, J.J.; Henninka, W.E. A new class of polymerizable dextrans with hydrolyzable groups: Hydroxyethyl methacrylated dextran with and without oligolactate spacer. Polymer 1997, 38, 6235–6242. [Google Scholar] [CrossRef]
- Liang, H.-F.; Chen, C.-T.; Chen, S.-C.; Kulkarni, A.R.; Chiu, Y.-L.; Chen, M.-C.; Sung, H.-W. Paclitaxel-loaded poly(γ-glutamic acid)-poly(lactide) nanoparticles as a targeted drug delivery system for the treatment of liver cancer. Biomaterials 2006, 27, 2051–2059. [Google Scholar] [CrossRef]
- Keresztessy, Z.; Bodnár, M.; Ber, E.; Hajdu, I.; Zhang, M.; Hartmann, J.F.; Minko, T.; Borbély, J. Self-assembling chitosan/poly-γ-glutamic acid nanoparticles for targeted drug delivery. Colloid Polym. Sci. 2009, 287, 759–765. [Google Scholar] [CrossRef]
- Lu, K.-Y.; Lin, C.-W.; Hsu, C.-H.; Ho, Y.-C.; Chuang, E.-Y.; Sung, H.-W.; Mi, F.-L. FRET-based dual-emission and pH-responsive nanocarriers for enhanced delivery of protein across intestinal epithelial cell barrier. ACS Appl. Mater. Interfaces 2014, 6, 18275–18289. [Google Scholar] [CrossRef]
- Liao, Z.-X.; Peng, S.-F.; Chiu, Y.-L.; Hsiao, C.-W.; Liu, H.-Y.; Lim, W.-H.; Lu, H.-M.; Sung, H.-W. Enhancement of efficiency of chitosan-based complexes for gene transfection with poly(γ-glutamic acid) by augmenting their cellular uptake and intracellular unpackage. J. Control. Release 2014, 193, 304–315. [Google Scholar] [CrossRef]
- Teixeira, G.Q.; Pereira, C.L.; Castro, F.; Ferreira, J.R.; Gomez-Lazaro, M.; Aguiar, P.; Barbosa, M.A.; Neidlinger-Wilke, C.; Goncalves, R.M. Anti-inflammatory Chitosan/Poly-γ-glutamic acid nanoparticles control inflammation while remodeling extracellular matrix in degenerated intervertebral disc. Acta Biomater. 2016, 42, 168–179. [Google Scholar] [CrossRef]
- Kim, E.S.; Lee, J.-S.; Lee, H.G. Nanoencapsulation of red ginseng extracts using chitosan with polyglutamic acid or fucoidan for improving antithrombotic activities. J. Agric. Food Chem. 2016, 64, 4765–4771. [Google Scholar] [CrossRef]
- Chen, M.-C.; Ling, M.-H.; Kusuma, S.J. Poly-γ-glutamic acid microneedles with a supporting structure design as a potential tool for transdermal delivery of insulin. Acta Biomater. 2015, 24, 106–116. [Google Scholar] [CrossRef]
- Yan, X.; Tong, Z.; Chen, Y.; Mo, Y.; Feng, H.; Li, P.; Qu, X.; Jin, S. Bioresponsive Materials for Drug Delivery Based on Carboxymethyl Chitosan/Poly(γ-Glutamic Acid) Composite Microparticles. Mar. Drugs 2017, 15, 127. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.P.; Shukla, T.; Dhote, V.K.; Mishra, D.K.; Maheshwari, R.; Tekade, R.K. Chapter 4—Use of Polymers in Controlled Release of Active Agents. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Academic Press: London, UK, 2019; pp. 113–172. [Google Scholar] [CrossRef]
- Staehlke, S.; Lehnfeld, J.; Schneider, A.; Nebe, J.B.; Müller, R. Terminal chemical functions of polyamidoamine dendrimer surfaces and its impact on bone cell growth. Mater. Sci. Eng. C 2019, 101, 190–203. [Google Scholar] [CrossRef]
- Busson, P.; Ihre, H.; Hult, A. Synthesis of a Novel Dendritic Liquid Crystalline Polymer Showing a Ferroelectric SmC* Phase. J. Am. Chem. Soc. 1998, 120, 9070–9071. [Google Scholar] [CrossRef]
- Knapen, J.W.J.; Van Der Made, A.W.; De Wilde, J.C.; Van Leeuwen, P.W.N.M.; Wijkens, P.; Grove, D.M.; Van Koten, G. Homogeneous catalysts based on silane dendrimers functionalized with arylnickel(II) complexes. Nature 1994, 372, 659–663. [Google Scholar] [CrossRef]
- Zhong, H.; He, Z.-G.; Li, Z.; Li, G.-Y.; Shen, S.-R.; Li, X.-L. Studies on Polyamidoamine Dendrimers as Efficient Gene Delivery Vector. J. Biomater. Appl. 2007, 22, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.X.; Redemann, C.T.; Szoka, F.C. In Vitro Gene Delivery by Degraded Polyamidoamine Dendrimers. Bioconj. Chem. 1996, 7, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Ito, Y.; Higashihara, T.; Ueda, M. Synthesis of a novel water-soluble polyamide dendrimer based on a facile convergent method. Eur. Polym. J. 2009, 45, 1994–2001. [Google Scholar] [CrossRef]
- Tomalia, D.A. Starburst Dendrimers—Molecular-Level Control of Size, Shape, Surface-Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Abstr. Pap. Am. Chem. Soc. 1990, 199, 315-ORGN. [Google Scholar] [CrossRef]
- Venditto, V.J.; Regino, C.A.S.; Brechbiel, M.W. PAMAM Dendrimer Based Macromolecules as Improved Contrast Agents. Mol. Pharm. 2005, 2, 302–311. [Google Scholar] [CrossRef]
- Florendo, M.; Figacz, A.; Srinageshwar, B.; Sharma, A.; Swanson, D.; Dunbar, G.L.; Rossignol, J. Use of Polyamidoamine Dendrimers in Brain Diseases. Molecules 2018, 23, 2238. [Google Scholar] [CrossRef] [Green Version]
- Devarakonda, B.; Hill, R.A.; Liebenberg, W.; Brits, M.; de Villiers, M.M. Comparison of the aqueous solubilization of practically insoluble niclosamide by polyamidoamine (PAMAM) dendrimers and cyclodextrins. Int. J. Pharm. 2005, 304, 193–209. [Google Scholar] [CrossRef]
- Samad, A.; Alam, M.I.; Saxena, K. Dendrimers: A class of polymers in the nanotechnology for the delivery of active pharmaceuticals. Curr. Pharm. Des. 2009, 15, 2958–2969. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.H.; El-Sayed, M.E.H. Dendrimers as Carriers for Delivery of Chemotherapeutic Agents. Chem. Rev. 2009, 109, 3141–3157. [Google Scholar] [CrossRef]
- Dong, R.; Zhou, Y.; Zhu, X. Supramolecular Dendritic Polymers: From Synthesis to Applications. Acc. Chem. Res. 2014, 47, 2006–2016. [Google Scholar] [CrossRef]
- Li, J.; Liang, H.; Liu, J.; Wang, Z. Poly (amidoamine) (PAMAM) dendrimer mediated delivery of drug and pDNA/siRNA for cancer therapy. Int. J. Pharm. 2018, 546, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Bahadır, E.B.; Sezgintürk, M.K. Poly (amidoamine)(PAMAM): An emerging material for electrochemical bio (sensing) applications. Talanta 2016, 148, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.; Liu, N.; Guo, N.; Gao, C.; Hao, Y.; Chen, L.; Zhang, X. In vitro studies of phospholipid-modified PAMAM-siMDR1 complexes for the reversal of multidrug resistance in human breast cancer cells. Int. J. Pharm. 2017, 530, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Javar, R.A.; Bin Noordin, M.I.; Khoobi, M.; Ghaedi, A. Fatty Acid Based Polyamide for Application in Drug Delivery System: Synthesis, Characterization, Drug Loading and In Vitro Drug Release Study. J. Inorg. Organomet. Polym. Mater. 2020, 30, 2520–2532. [Google Scholar] [CrossRef]
- Adeleke, O.A. In vitro characterization of a synthetic polyamide-based erodible compact disc for extended drug release. SN Appl. Sci. 2020, 2, 2152. [Google Scholar] [CrossRef]
- Salahuddin, N.; Elbarbary, A.; Allam, N.G.; Hashim, A.F. 5-Phenyl-1,3,4-oxadiazole-2-thiol/polyamide-montmorillonite microbicides nanocomposites as drug delivery system. J. Phys. Org. Chem. 2018, 31, e3834. [Google Scholar] [CrossRef]
- Yavvari, P.S.; Gupta, S.; Arora, D.; Nandicoori, V.; Srivastava, A.; Bajaj, A. Clathrin-Independent Killing of Intracellular Mycobacteria and Biofilm Disruptions Using Synthetic Antimicrobial Polymers. Biomacromolecules 2017, 18, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Menezes, P.D.P.; Frank, L.A.; Lima, B.D.S.; Carvalho, Y.; Serafini, M.R.; Quintans-Júnior, L.J.; Pohlmann, A.R.; Guterres, S.; Araújo, A.A.D.S. Hesperetin-loaded lipid-core nanocapsules in polyamide: A new textile formulation for topical drug delivery. Int. J. Nanomed. 2017, 12, 2069–2079. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Chen, L.; Zhou, M.; Li, J.; Liu, J.; Fang, H.; Zeng, Y.; Sun, J.; Wang, Z. Multi-Layered Polyamide/Collagen Scaffolds with Topical Sustained Release of N-Acetylcysteine for Promoting Wound Healing. Int. J. Nanomed. 2020, 15, 1349–1361. [Google Scholar] [CrossRef] [Green Version]
- Boddu, S.H.; Jwala, J.; Chowdhury, M.R.; Mitra, A.K. In Vitro Evaluation of a Targeted and Sustained Release System for Retinoblastoma Cells Using Doxorubicin as a Model Drug. J. Ocul. Pharmacol. Ther. 2010, 26, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.Y.; Bae, Y.H. Polymer Architecture and Drug Delivery. Pharm. Res. 2006, 23, 1–30. [Google Scholar] [CrossRef]
- Cabral, H.; Kataoka, K. Progress of drug-loaded polymeric micelles into clinical studies. J. Control. Release 2014, 190, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2012, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khayat, D.; Antoine, E.-C.; Coeffic, D. Taxol in the management of cancers of the breast and the ovary. Cancer Investig. 2000, 18, 242–260. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Rajabi, M.; Mousa, S.A. Multifunctional Nanomaterials and Their Applications in Drug Delivery and Cancer Therapy. Nanomaterials 2015, 5, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B.; Donehower, R.; Wiernik, P.; Ohnuma, T.; Gralla, R.; Trump, D.; Baker Jr, J.; Van Echo, D.; Von Hoff, D.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (taxol). N. Engl. J. Med. 1995, 332, 1004–1014. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Chaudhry, V.; Forastiere, A.A.; Sartorius, S.E.; Ettinger, D.S.; Grochow, L.B.; Lubejko, B.G.; Cornblath, D.R.; Donehower, R.C. Phase I and pharmacologic study of paclitaxel and cisplatin with granulocyte colony-stimulating factor: Neuromuscular toxicity is dose-limiting. J. Clin. Oncol. 1993, 11, 2010–2020. [Google Scholar] [CrossRef]
- Wasserheit, C.; Frazein, A.; Oratz, R.; Sorich, J.; Downey, A.; Hochster, H.; Chachoua, A.; Wernz, J.; Zeleniuch-Jacquotte, A.; Blum, R.; et al. Phase II trial of paclitaxel and cisplatin in women with advanced breast cancer: An active regimen with limiting neurotoxicity. J. Clin. Oncol. 1996, 14, 1993–1999. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Matsumura, Y.; Suzuki, M.; Shimizu, K.; Goda, R.; Nakamura, I.; Nakatomi, I.; Yokoyama, M.; Kataoka, K.; Kakizoe, T. NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br. J. Cancer 2005, 92, 1240–1246. [Google Scholar] [CrossRef] [Green Version]
- Schlechter, B.; Neumann, A.; Wilchek, M.; Arnon, R. Soluble polymers as carriers of cis-platinum. J. Control. Release 1989, 10, 75–87. [Google Scholar] [CrossRef]
- Kazunori, K.; Kwon, G.S.; Masayuki, Y.; Teruo, O.; Yasuhisa, S. Block copolymer micelles as vehicles for drug delivery. J. Control. Release 1993, 24, 119–132. [Google Scholar] [CrossRef]
- Masayuki, Y.; Mizue, M.; Noriko, Y.; Teruo, O.; Yasuhisa, S.; Kazunori, K.; Shohei, I. Polymer micelles as novel drug carrier: Adriamycin-conjugated poly(ethylene glycol)-poly(aspartic acid) block copolymer. J. Control. Release 1990, 11, 269–278. [Google Scholar] [CrossRef]
- Yokoyama, M.; Okano, T.; Sakurai, Y.; Ekimoto, H.; Shibazaki, C.; Kataoka, K. Toxicity and antitumor activity against solid tumors of micelle-forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res. 1991, 51, 3229–3236. [Google Scholar] [PubMed]
- Matsumura, Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv. Drug Deliv. Rev. 2008, 60, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Negishi, T.; Koizumi, F.; Uchino, H.; Kuroda, J.; Kawaguchi, T.; Naito, S.; Matsumura, Y. NK105, a paclitaxel-incorporating micellar nanoparticle, is a more potent radiosensitising agent compared to free paclitaxel. Br. J. Cancer 2006, 95, 601–606. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Kato, K.; Yasui, H.; Morizane, C.; Ikeda, M.; Ueno, H.; Muro, K.; Yamada, Y.; Okusaka, T.; Shirao, K.; et al. A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation. Br. J. Cancer 2007, 97, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Chin, K.; Yoshikawa, T.; Yamaguchi, K.; Tsuji, Y.; Esaki, T.; Sakai, K.; Kimura, M.; Hamaguchi, T.; Shimada, Y.; et al. Phase II study of NK105, a paclitaxel-incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer. Investig. New Drugs 2011, 30, 1621–1627. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Mukai, H.; Saeki, T.; Ro, J.; Lin, Y.-C.; Nagai, S.E.; Lee, K.S.; Watanabe, J.; Ohtani, S.; Kim, S.B.; et al. A multi-national, randomised, open-label, parallel, phase III non-inferiority study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients. Br. J. Cancer 2019, 120, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Oprita, A.; Sevastre, A.-S. New pharmaceutical dosage forms used in the treatment of breast cancer. polymeric micelles. Med. Oncol. 2020, 1, 38–52. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Tsuji, A.; Yamaguchi, K.; Takeda, K.; Uetake, H.; Esaki, T.; Amagai, K.; Sakai, D.; Baba, H.; Kimura, M.; et al. A phase II study of NK012, a polymeric micelle formulation of SN-38, in unresectable, metastatic or recurrent colorectal cancer patients. Cancer Chemother. Pharmacol. 2018, 82, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.A.V. Π-π Stacked Polymeric Micelles as Carriers of Anticancer Therapeutics for Hematological Malignancies. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Horwich, A.; Sleijfer, D.T.; Fosså, S.; Kaye, S.; Oliver, R.; Cullen, M.; Mead, G.; de Wit, R.; De Mulder, P.; Dearnaley, D. Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good-prognosis metastatic nonseminomatous germ cell cancer: A Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J. Clin. Oncol. 1997, 15, 1844–1852. [Google Scholar]
- Brenner, B.; Tang, L.H.; Shia, J.; Klimstra, D.S.; Kelsen, D.P. Small Cell Carcinomas of the Gastrointestinal Tract: Clinicopathological Features and Treatment Approach. Semin. Oncol. 2007, 34, 43–50. [Google Scholar] [CrossRef]
- Screnci, D.; Mckeage, M.; Galettis, P.; Hambley, T.W.; Palmer, B.D.; Baguley, B.C. Relationships between hydrophobicity, reactivity, accumulation and peripheral nerve toxicity of a series of platinum drugs. Br. J. Cancer 2000, 82, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Cleare, M.; Hydes, P.; Malerbi, B.; Watkins, D. Anti-tumour platinum complexes: Relationships between chemical properties and activity. Biochimie 1978, 60, 835–850. [Google Scholar] [CrossRef]
- Du Bois, A.; Lück, H.-J.; Meier, W.; Adams, H.-P.; Möbus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schröder, W. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Natl. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.; Tabernero, J.; Twelves, C.; Brunet, R.; Butts, C.; Conroy, T.; DeBraud, F.; Figer, A.; Grossmann, J.; Sawada, N.; et al. XELOX (Capecitabine Plus Oxaliplatin): Active First-Line Therapy for Patients with Metastatic Colorectal Cancer. J. Clin. Oncol. 2004, 22, 2084–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volovat, S.R.; Ciuleanu, T.-E.; Koralewski, P.; Olson, J.E.G.; Croitoru, A.; Koynov, K.; Stabile, S.; Cerea, G.; Osada, A.; Bobe, I.; et al. A multicenter, single-arm, basket design, phase II study of NC-6004 plus gemcitabine in patients with advanced unresectable lung, biliary tract, or bladder cancer. Oncotarget 2020, 11, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Bansal, K.; Sasso, L.; Makwana, H.; Awwad, S.; Brocchini, S.; Alexander, C. Nanopharmacy: Exploratory Methods for Polymeric Materials. In Pharmaceutical Nanotechnology: Innovation and Production; Wiley-VCH Verlag GmbH & Company KGaA: Weinheim, Germany, 2017; pp. 231–270. [Google Scholar] [CrossRef]
- Plummer, R.; Wilson, R.; Calvert, H.; Boddy, A.; Griffin, M.; Sludden, J.; Tilby, M.; Eatock, M.; Pearson, D.; Ottley, C.; et al. A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours. Br. J. Cancer 2011, 104, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Osada, A.; Mangel, L.; Fijuth, J.; Żurawski, B.; Ursulovic, T.; Nikolin, B.; Djan, I.; Olson, J.G. Phase IIa/IIb clinical trial of NC-6004 (Nanoparticle Cisplatin) plus Pembrolizumab in patients with head and neck cancer (HNSCC) who have failed platinum or a platinum-containing regimen. Eur. J. Cancer 2020, 138, S35. [Google Scholar] [CrossRef]
- Shi, Y.; Lammers, T.; Storm, G.; Hennink, W.E. Physico-Chemical Strategies to Enhance Stability and Drug Retention of Polymeric Micelles for Tumor-Targeted Drug Delivery. Macromol. Biosci. 2016, 17, 1600160. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Xie, J.; Zhang, X.; Sun, W.; Zhao, H.; Li, Y.; Wang, C. An overview of polymeric nanomicelles in clinical trials and on the market. Chin. Chem. Lett. 2020, 32, 243–257. [Google Scholar] [CrossRef]
- Mukai, H.; Kogawa, T.; Matsubara, N.; Naito, Y.; Sasaki, M.; Hosono, A. A first-in-human Phase 1 study of epirubicin-conjugated polymer micelles (K-912/NC-6300) in patients with advanced or recurrent solid tumors. Investig. New Drugs 2017, 35, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Abbina, S.; Parambath, A. PEGylation and its alternatives: A summary. In Engineering of Biomaterials for Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2018; pp. 363–376. [Google Scholar]
- Doppalapudi, S.; Jain, A.; Khan, W.; Domb, A.J. Biodegradable polymers—An overview. Polym. Adv. Technol. 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Sill, K.N.; Sullivan, B.; Carie, A.; Semple, J.E. Synthesis and Characterization of Micelle-Forming PEG-Poly(Amino Acid) Copolymers with Iron-Hydroxamate Cross-Linkable Blocks for Encapsulation and Release of Hydrophobic Drugs. Biomacromolecules 2017, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, S.C.; Chan, D.P.; Shoichet, M.S. Polymeric micelle stability. Nano Today 2012, 7, 53–65. [Google Scholar] [CrossRef]
- Letchford, K.; Liggins, R.; Wasan, K.M.; Burt, H. In vitro human plasma distribution of nanoparticulate paclitaxel is dependent on the physicochemical properties of poly(ethylene glycol)-block-poly(caprolactone) nanoparticles. Eur. J. Pharm. Biopharm. 2009, 71, 196–206. [Google Scholar] [CrossRef]
- Opanasopit, P.; Yokoyama, M.; Watanabe, M.; Kawano, K.; Maitani, Y.; Okano, T. Influence of serum and albumins from different species on stability of camptothecin-loaded micelles. J. Control. Release 2005, 104, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Poon, Z.; Engler, A.C.; Bonner, D.K.; Hammond, P.T. Enhanced Stability of Polymeric Micelles Based on Postfunctionalized Poly(ethylene glycol)-b-poly(γ-propargyl l-glutamate): The Substituent Effect. Biomacromolecules 2012, 13, 1315–1322. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.O.; Oberoi, H.S.; Desale, S.; Kabanov, A.; Bronich, T.K. Polypeptide nanogels with hydrophobic moieties in the cross-linked ionic cores: Synthesis, characterization and implications for anticancer drug delivery. J. Drug Target. 2013, 21, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Ohya, Y.; Takeda, S.; Shibata, Y.; Ouchi, T.; Kano, A.; Iwata, T.; Mochizuki, S.; Taniwaki, Y.; Maruyama, A. Evaluation of polyanion-coated biodegradable polymeric micelles as drug delivery vehicles. J. Control. Release 2011, 155, 104–110. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Polymer |
|---|---|
| Natural Polymers | |
| Protein–based polymers | Collagen, albumin, gelatin |
| Polysaccharides | Agarose, alginate, carrageenan, hyaluronic acid, dextran, chitosan, cyclodextrins |
| Synthetic polymers | |
| Biodegradable | |
| Polyesters | Poly(lactic acid), poly(glycolic acid), poly(hydroxy butyrate), poly(ε-caprolactone), poly(β-malic acid), poly(dioxanes) |
| Polyanhydrides | Poly(sebacic acid), poly(adipic acid), poly(terphthalic acid) and various copolymers |
| Polyamides | Poly(imino carbonates), polyamino acids |
| Phosphorus-based polymers | Polyphosphates, polyphosphonates, polyphosphazenes |
| Others | Poly(cyano acrylates), polyurethanes, polyortho esters, polydihydropyrans, polyacetals |
| Non-biodegradable polymers | |
| Acrylic polymers | Polymethacrylates, poly(methyl methacrylate), poly hydro(ethyl methacrylate) |
| Cellulose derivatives | Carboxymethyl cellulose, ethyl cellulose, cellulose acetate, cellulose acetate propionate, hydroxyl propyl methyl cellulose |
| Silicones | Polydimethyl siloxane, colloidal silica |
| Others | Polyvinyl pyrrolidone, ethyl vinyl acetate, poloxamers, poloxamines |
| Polymer | Structure | Applications |
|---|---|---|
| Poly(lysine) |  |
|
| Poly(glutamic acid) |  |
|
| Poly(α-L-aspartic acid) |  |
|
| Polyamidoamine (G1) |  |
|
| Treatment | Dose (mg/kg) | C5 min (µg/mL) | t1/2 z (h) | AUC0-t (µg·h/mL) | AUC0-inf. (µg·h/mL) | Cltot (mL/h·kg) | Vss (L/kg) |
|---|---|---|---|---|---|---|---|
| Plasma PTX | 50 | 59.32 | 0.98 | 90.2 a | 91.3 | 547.6 | 684.6 |
| PTX | 100 | 157.67 | 1.84 | 309.0 b | 309.0 | 323.6 | 812.2 |
| NK105 | 50 | 1157.03 | 5.99 | 7860.9 c | 7862.3 | 6.4 | 46.4 |
| NK105 | 100 | 1812.37 | 6.82 | 15,565.7 c | 15,573.6 | 6.4 | 54.8 |
| Treatment | Dose (mg/kg) | Cmax (µg/mL) | Tman (h) | t1/2 z (h) | AUC0-t (µg·h/mL) | AUC0-inf. (µg·h/mL) | |
| Tumor PTX | 50 | 12.50 | 2.0 | 7.02 | 120.8 b | 133.0 | |
| PTX | 100 | 28.57 | 0.5 | 8.06 | 330.4 c | 331.0 | |
| NK105 | 50 | 42.45 | 24.0 | 35.07 | 2360.1 c | 3192.0 | |
| NK105 | 100 | 71.09 | 6.0 | 73.66 | 3884.9 c | 7964.5 |
| Compound | Rat | Tmax a (h) | Cmax a (µg/mL) | t1/2 z (h) | AUC0-t (µg·h/mL) | AUC0-inf. (µg·h/mL) | CLtot (mL/h·kg) | MRT0-inf. (h) | Vss (L/kg) |
|---|---|---|---|---|---|---|---|---|---|
| CDDP | Mean s.d. | 0.083 | 11.67 0.57 | 34.50 16.14 | 20.47 2.25 | 75.73 26.13 | 70.67 20.34 | 46.57 22.38 | 3.00 0.61 |
| NC-6004 | Mean s.d. | 0.50 | 89.90 4.29 | 6.43 0.55 | 1325.90 77.85 | 1335.47 75.99 | 3.77 0.21 | 10.67 0.15 | 0.04 0.0023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boddu, S.H.S.; Bhagav, P.; Karla, P.K.; Jacob, S.; Adatiya, M.D.; Dhameliya, T.M.; Ranch, K.M.; Tiwari, A.K. Polyamide/Poly(Amino Acid) Polymers for Drug Delivery. J. Funct. Biomater. 2021, 12, 58. https://doi.org/10.3390/jfb12040058
Boddu SHS, Bhagav P, Karla PK, Jacob S, Adatiya MD, Dhameliya TM, Ranch KM, Tiwari AK. Polyamide/Poly(Amino Acid) Polymers for Drug Delivery. Journal of Functional Biomaterials. 2021; 12(4):58. https://doi.org/10.3390/jfb12040058
Chicago/Turabian StyleBoddu, Sai H. S., Prakash Bhagav, Pradeep K. Karla, Shery Jacob, Mansi D. Adatiya, Tejas M. Dhameliya, Ketan M. Ranch, and Amit K. Tiwari. 2021. "Polyamide/Poly(Amino Acid) Polymers for Drug Delivery" Journal of Functional Biomaterials 12, no. 4: 58. https://doi.org/10.3390/jfb12040058
APA StyleBoddu, S. H. S., Bhagav, P., Karla, P. K., Jacob, S., Adatiya, M. D., Dhameliya, T. M., Ranch, K. M., & Tiwari, A. K. (2021). Polyamide/Poly(Amino Acid) Polymers for Drug Delivery. Journal of Functional Biomaterials, 12(4), 58. https://doi.org/10.3390/jfb12040058