

Optimization of Tyrosine Kinase Inhibitor-Loaded Gold Nanoparticles for Stimuli-Triggered Antileukemic Drug Release
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis and Biofunctionalization of MDS-Loaded Gold Nanoparticles (MDS-GNP)
2.2.2. Evaluation of the Drug Release Profile
2.2.3. Particle Characterization
2.2.4. Cell Culture Protocol
2.2.5. XTT Assay
2.2.6. Cell Preparation for Dark Field Microscopy
2.2.7. Microscopic Analysis
3. Results and Discussion
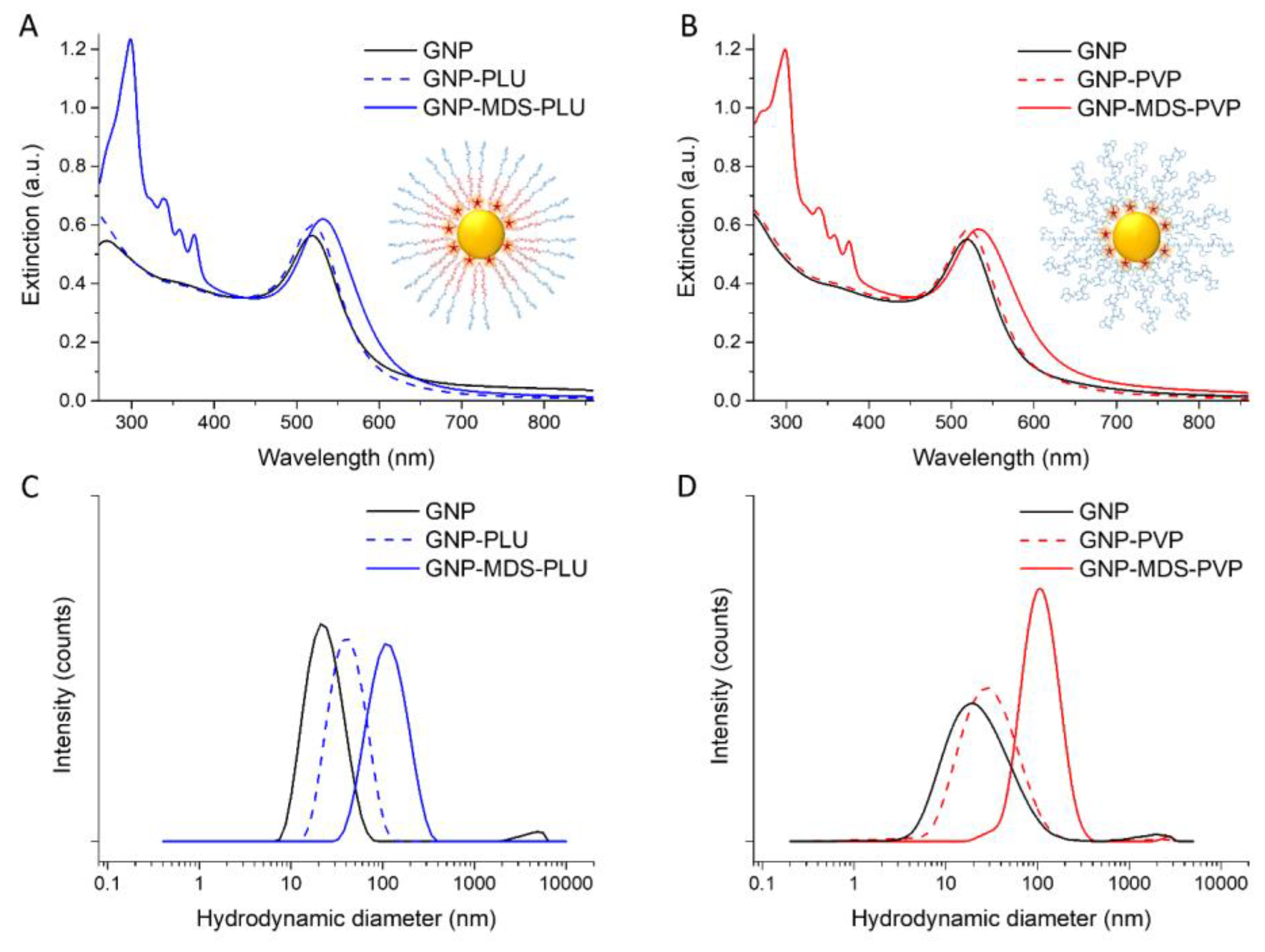
3.1. Fabrication and Characterization of Drug-Loaded Gold Nanoparticles
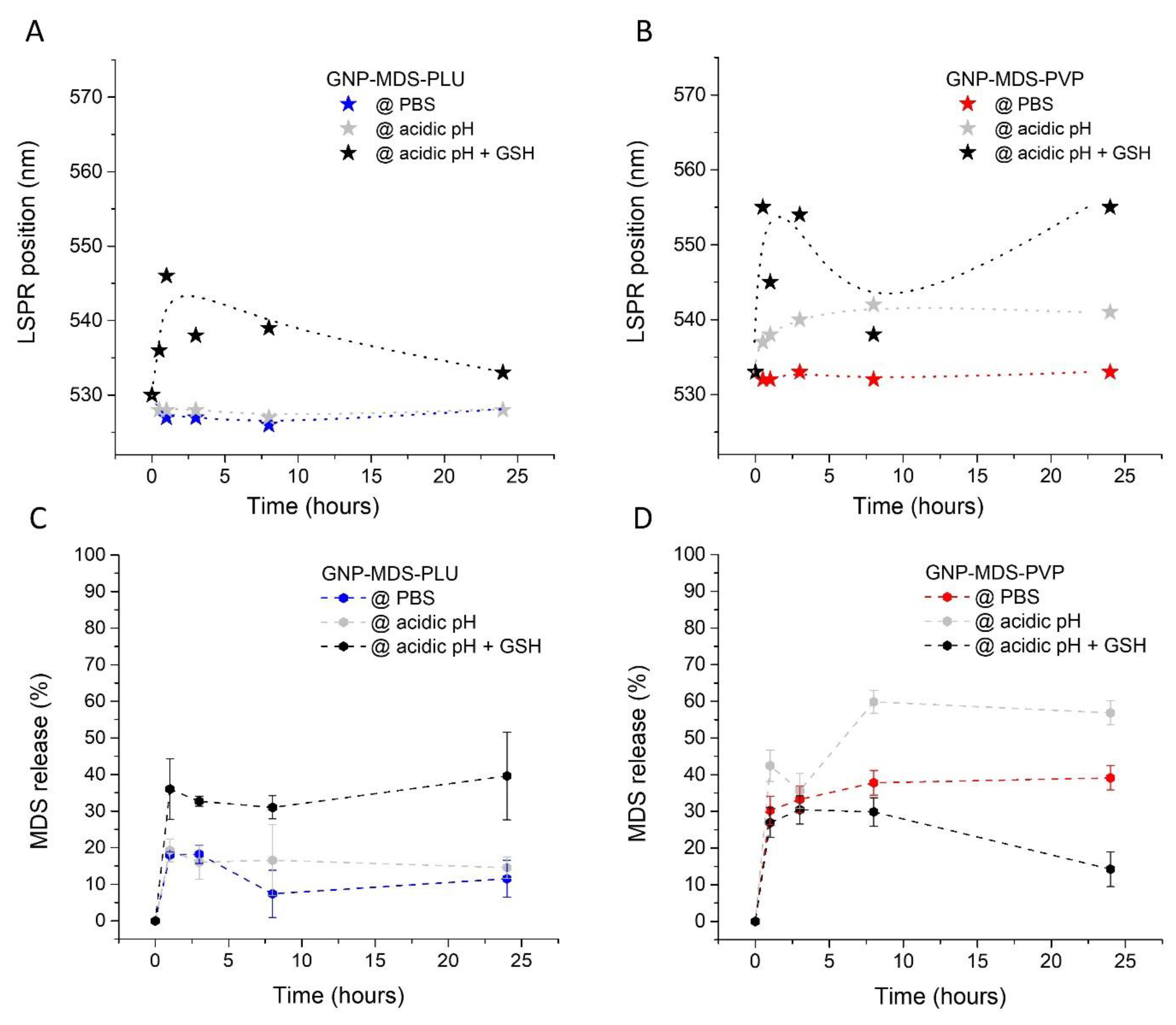
3.2. Evaluation of the Drug Release Profile
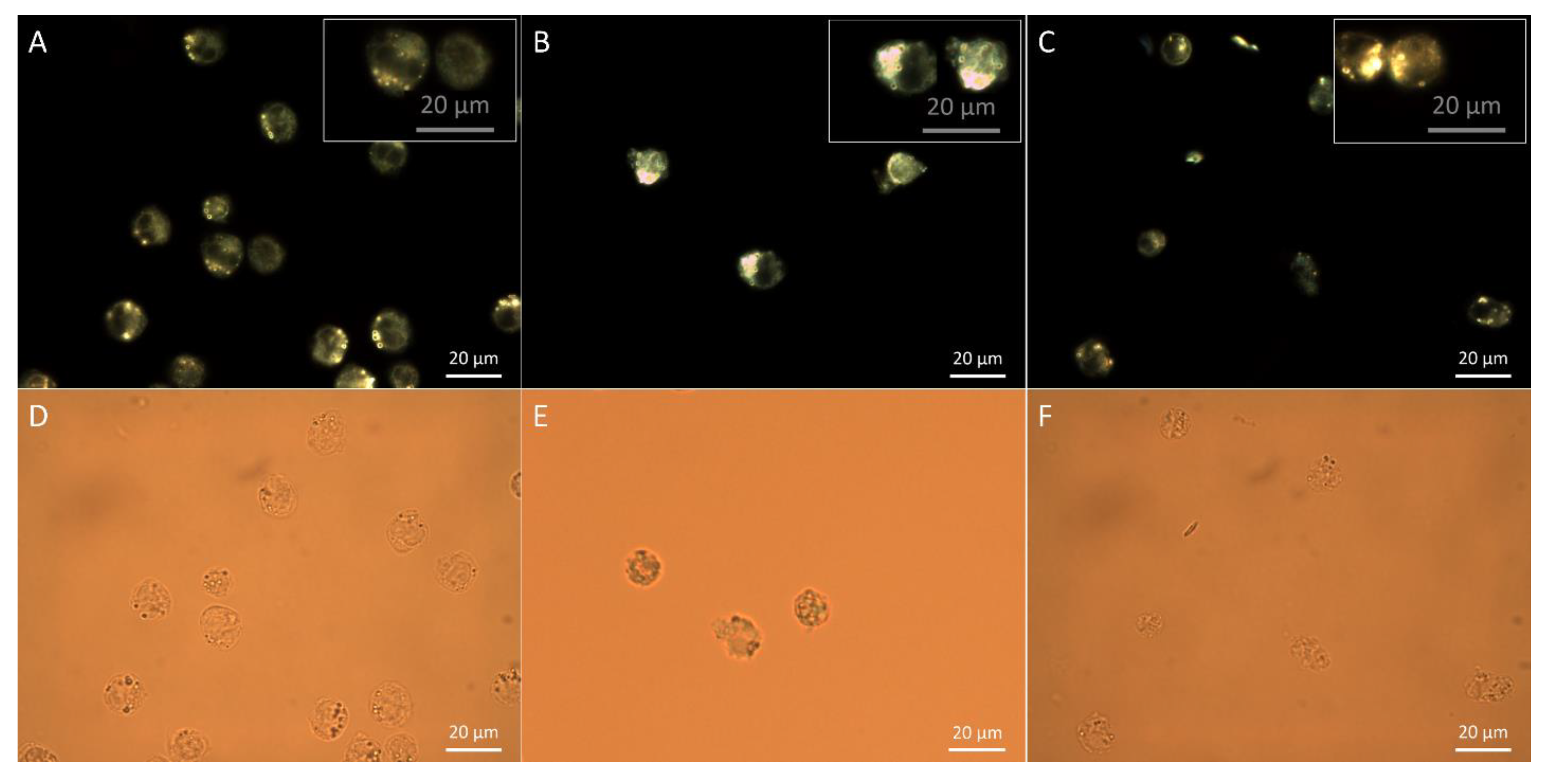
3.3. Assessment of Particle Internalization by Dark Field Microscopy Imaging
3.4. Cellular Toxicity
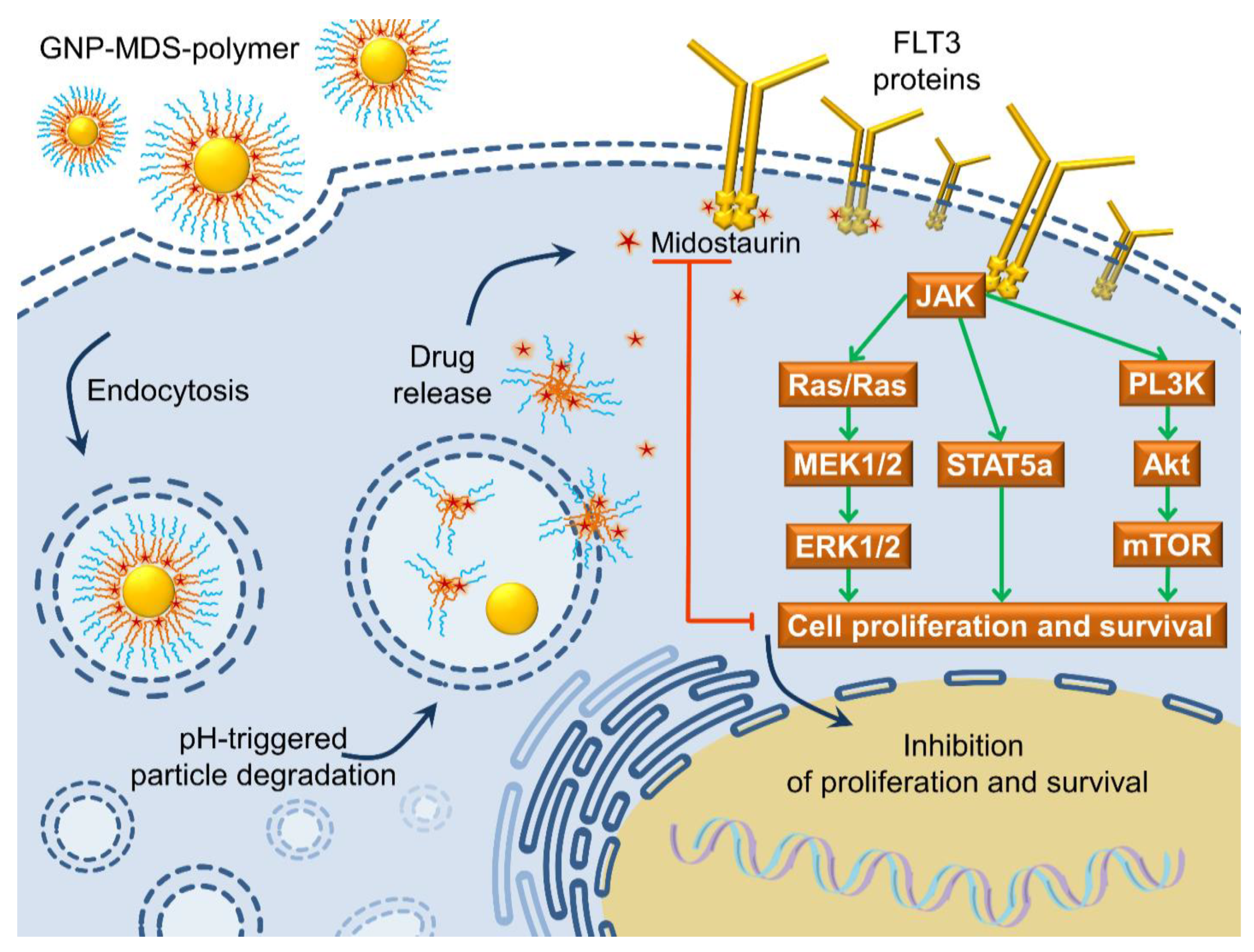
3.5. The FLT3 Signaling Pathway as a Pharmacological Target: Mechanisms of Drug and Drug-Nanoparticle Compounds Action
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Visser, O.; Trama, A.; Maynadié, M.; Stiller, C.; Marcos-Gragera, R.; De Angelis, R.; Mallone, S.; Tereanu, C.; Allemani, C.; Ricardi, U.; et al. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur. J. Cancer 2012, 48, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [Green Version]
- Key Statistics for Acute Myeloid Leukemia (AML)’. Available online: https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html (accessed on 14 September 2021).
- Dong, Y.; Shi, O.; Zeng, Q.; Lu, X.; Wang, W.; Li, Y.; Wang, Q. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017. Exp. Hematol. Oncol. 2020, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Petrushev, B.; Boca, S.; Simon, T.; Berce, C.; Frinc, I.; Dima, D.; Selicean, S.; Gafencu, G.A.; Tanase, A.; et al. Gold nanoparticles enhance the effect of tyrosine kinase inhibitors in acute myeloid leukemia therapy. Int. J. Nanomed. 2016, 11, 641–660. [Google Scholar] [CrossRef] [Green Version]
- Megías-Vericat, J.E.; Ballesta-López, O.; Barragán, E.; Martínez-Cuadrón, D.; Montesinos, P. Tyrosine kinase inhibitors for acute myeloid leukemia: A step toward disease control? Blood Rev. 2020, 44, 100675. [Google Scholar] [CrossRef]
- Fleuren, E.; Zhang, L.; Wu, J.; Daly, R. The kinome ‘at large’ in cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, S.; Desplat, V.; Villacreces, A.; Guitart, A.V.; Milpied, N.; Pigneux, A.; Vigon, I.; Pasquet, J.-M.; Dumas, P.-Y. Targeting Tyrosine Kinases in Acute Myeloid Leukemia: Why, Who and How? Int. J. Mol. Sci. 2019, 20, 3429. [Google Scholar] [CrossRef] [Green Version]
- Kottaridis, P.D.; Gale, R.E.; Linch, D.C. Flt3 mutations and leukaemia. Br. J. Haematol. 2003, 122, 523–538. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Tomuleasa, C.; Bojan, A.; Berindan-Neagoe, I.; Boca, S.; Astilean, S. Design of FLT3 Inhibitor—Gold Nanoparticle Conjugates as Potential Therapeutic Agents for the Treatment of Acute Myeloid Leukemia. Nanoscale Res. Lett. 2015, 10, 466. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Deshantri, A.K.; Moreira, A.V.; Ecker, V.; Mandhane, S.N.; Schiffelers, R.M.; Buchner, M.; Fens, M.H. Nanomedicines for the treatment of hematological malignancies. J. Control. Release 2018, 287, 194–215. [Google Scholar] [CrossRef]
- Zhu, Y.; Liao, L. Applications of Nanoparticles for Anticancer Drug Delivery: A Review. J. Nanosci. Nanotechnol. 2015, 15, 4753–4773. [Google Scholar] [CrossRef]
- Liao, J.; Jia, Y.; Wu, Y.; Shi, K.; Yang, D.; Li, P.; Qian, Z. Physical-, chemical-, and biological-responsive nanomedicine for cancer therapy. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1581. [Google Scholar] [CrossRef]
- Rao, N.V.; Ko, H.; Lee, J.; Park, J.H. Recent Progress and Advances in Stimuli-Responsive Polymers for Cancer Therapy. Front. Bioeng. Biotechnol. 2018, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- Iacovita, C.; Stiufiuc, R.; Radu, T.; Florea, A.; Stiufiuc, G.; Dutu, A.; Mican, S.; Tetean, R.; Lucaciu, C.M. Polyethylene Glycol-Mediated Synthesis of Cubic Iron Oxide Nanoparticles with High Heating Power. Nanoscale Res. Lett. 2015, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Tatar, A.-S.; Jurj, A.; Tomuleasa, C.; Florea, A.; Berindan-Neagoe, I.; Cialla-May, D.; Popp, J.; Astilean, S.; Boca, S. CD19-targeted, Raman tagged gold nanourchins as theranostic agents against acute lymphoblastic leukemia. Colloids Surf. B Biointerfaces 2019, 184, 110478. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Simon, T.; Tatar, A.-S.; Craciun, A.-M.; Vulpoi, A.; Jurj, M.-A.; Florea, A.; Tomuleasa, C.; Berindan-Neagoe, I.; Astilean, S.; Boca, S. Antibody Conjugated, Raman Tagged Hollow Gold–Silver Nanospheres for Specific Targeting and Multimodal Dark-Field/SERS/Two Photon-FLIM Imaging of CD19(+) B Lymphoblasts. ACS Appl. Mater. Interfaces 2017, 9, 21155–21168. [Google Scholar] [CrossRef] [PubMed]
- Gharatape, A.; Salehi, R. Recent progress in theranostic applications of hybrid gold nanoparticles. Eur. J. Med. Chem. 2017, 138, 221–233. [Google Scholar] [CrossRef]
- Taghizadeh, B.; Taranejoo, S.; Monemian, S.A.; Moghaddam, Z.S.; Daliri, K.; Derakhshankhah, H.; Derakhshani, Z. Classification of stimuli–responsive polymers as anticancer drug delivery systems. Drug Deliv. 2015, 22, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Han, W.; Zhao, X.; Tang, W.; Wang, F. Epirubicin-loaded marine carrageenan oligosaccharide capped gold nanoparticle system for pH-triggered anticancer drug release. Sci. Rep. 2019, 9, 6754. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, B. Preparation and characterization of polymeric pH-sensitive STEALTH® nanoparticles for tumor delivery of a lipophilic prodrug of paclitaxel. Int. J. Pharm. 2011, 408, 208–212. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Ding, Y.; Yan, W. Oxidized phospholipid based pH sensitive micelles for delivery of anthracyclines to resistant leukemia cells in vitro. Int. J. Pharm. 2012, 422, 409–417. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Liu, Z.; Liu, X.; Chen, L.; Yan, W. Bioactive Lipids-Based pH Sensitive Micelles for Co-Delivery of Doxorubicin and Ceramide to Overcome Multidrug Resistance in Leukemia. Pharm. Res. 2013, 30, 2902–2916. [Google Scholar] [CrossRef]
- Lin, W.; Yao, N.; Qian, L.; Zhang, X.; Chen, Q.; Wang, J.; Zhang, L. pH-responsive unimolecular micelle-gold nanoparticles-drug nanohybrid system for cancer theranostics. Acta Biomater. 2017, 58, 455–465. [Google Scholar] [CrossRef]
- Xiong, D.; Zhang, X.; Peng, S.; Gu, H.; Zhang, L. Smart pH-sensitive micelles based on redox degradable polymers as DOX/GNPs carriers for controlled drug release and CT imaging. Colloids Surf. B Biointerfaces 2018, 163, 29–40. [Google Scholar] [CrossRef]
- Xu, W.; Hong, Y.; Song, A.; Hao, J. Peptide-assembled hydrogels for pH-controllable drug release. Colloids Surf. B Biointerfaces 2020, 185, 110567. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lang, T.; Cun, D.; Zheng, Z.; Huang, Y.; Yin, Q.; Yu, H.; Li, Y. pH-Sensitive Nano-Complexes Overcome Drug Resistance and Inhibit Metastasis of Breast Cancer by Silencing Akt Expression. Theranostics 2017, 7, 4204–4216. [Google Scholar] [CrossRef]
- Sarkar, S.; Konar, S.; Prasad, P.N.; Rajput, S.; Kumar, B.N.P.; Rao, R.R.; Pathak, A.; Fisher, P.B.; Mandal, M. Micellear Gold Nanoparticles as Delivery Vehicles for Dual Tyrosine Kinase Inhibitor ZD6474 for Metastatic Breast Cancer Treatment. Langmuir 2017, 33, 7649–7659. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-T.; Lin, H.; Wang, C.-S.; Chang, C.-H.; Lin, A.M.-Y.; Yang, J.C.-H.; Lo, Y.-L. Improving the anticancer effect of afatinib and microRNA by using lipid polymeric nanoparticles conjugated with dual pH-responsive and targeting peptides. J. Nanobiotechnol. 2019, 17, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdagi, S.I.; Yildiz, U. Diosgenin-conjugated PCL–MPEG polymeric nanoparticles for the co-delivery of anticancer drugs: Design, optimization, in vitro drug release and evaluation of anticancer activity. New J. Chem. 2019, 43, 6622–6635. [Google Scholar] [CrossRef]
- Cortese, B.; D’Amone, S.; Gigli, G.; Palamà, I.E. Sustained anti-BCR-ABL activity with pH responsive imatinib mesylate loaded PCL nanoparticles in CML cells. MedChemComm 2015, 6, 212–221. [Google Scholar] [CrossRef]
- Cortese, B.; D’amone, S.; Palamà, I.E. Wool-Like Hollow Polymeric Nanoparticles for CML Chemo-Combinatorial Therapy. Pharmaceutics 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Suarasan, S.; Simon, T.; Boca, S.; Tomuleasa, C.; Astilean, S. Gelatin Coated Gold Nanoparticles as Carriers of FLT3 Inhibitors for Acute Myeloid Leukemia Treatment. Chem. Biol. Drug Des. 2016, 87, 927–935. [Google Scholar] [CrossRef]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Coskunpinar, E.; Anak, S.; Agaoglu, L.; Unuvar, A.; Devecioglu, O.; Aydogan, G.; Timur, C.; Oner, A.F.; Yildirmak, Y.; Celkan, T.; et al. Analysis of Chromosomal Aberrations and FLT3 gene Mutations in Childhood Acute Myelogenous Leukemia Patients. Turk. J. Hematol. 2012, 29, 225–232. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Frens, G. Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Fang, Y.; Wang, W. Investigation of pseudo-polyanion formation between polyvinylpyrrolidone and sodium dodecanoate in aqueous solution by capillary electrophoresis, conductometry, tensiometry and calcium stability. RSC Adv. 2017, 7, 9338–9346. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-H.; Ahn, J.; Shi, S.; Qin, D. Understanding the Role of Poly(vinylpyrrolidone) in Stabilizing and Capping Colloidal Silver Nanocrystals. ACS Nano 2021, 15, 14242–14252. [Google Scholar] [CrossRef]
- Abshire, C.; Murad, H.Y.; Arora, J.S.; Liu, J.; Mandava, S.H.; John, V.T.; Khismatullin, D.B.; Lee, B.R. Focused Ultrasound–Triggered Release of Tyrosine Kinase Inhibitor From Thermosensitive Liposomes for Treatment of Renal Cell Carcinoma. J. Pharm. Sci. 2017, 106, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Boca-Farcau, S.; Gabudean, A.-M.; Baldeck, P.; Astilean, S. LED-activated methylene blue-loaded Pluronic-nanogold hybrids for in vitro photodynamic therapy: Methylene blue-nanogold hybrids for LED induced PDT. J. Biophotonics 2013, 6, 950–959. [Google Scholar] [CrossRef]
- Verebes, G.S.; Melchiorre, M.; Garcia-Leis, A.; Ferreri, C.; Marzetti, C.; Torreggiani, A. Hyperspectral enhanced dark field microscopy for imaging blood cells. J. Biophotonics 2013, 6, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Stirewalt, D.L.; Radich, J.P. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Inhibition of the MEK/MAPK signal transduction pathway strongly impairs the growth of Flt3-ITD cells. Am. J. Hematol. 2006, 81, 154–155. [Google Scholar] [CrossRef]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Harigae, H.; Kaku, M.; Sasaki, T.; Licht, J.D. Flt3 mutation activates p21WAF1/CIP1 gene expression through the action of STAT5. Biochem. Biophys. Res. Commun. 2004, 316, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.-T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, H.; Miller, M. Midostaurin: An emerging treatment for acute myeloid leukemia patients. J. Blood Med. 2016, 7, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Saygin, C.; Carraway, H.E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Sattler, M.; Manley, P.W.; Griffin, J.D. Spotlight on midostaurin in the treatment of FLT3-mutated acute myeloid leukemia and systemic mastocytosis: Design, development, and potential place in therapy. OncoTargets Ther. 2017, 11, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Knapper, S.; Mills, K.I.; Gilkes, A.F.; Austin, S.J.; Walsh, V.; Burnett, A.K. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: The induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood 2006, 108, 3494–3503. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.; Freeman, C.; Giles, F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia 2012, 26, 2176–2185. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDD (nm) | PDI | Zeta-Potential (mV) | LSPR (nm) | LE (%) | MDS Concentration (µg/mL) | |
|---|---|---|---|---|---|---|
| GNP | 24.94 ± 3.8 | 0.525 | −61.6 ± 1.1 | 518 | - | - |
| GNP-PLU | 44.28 ± 0.29 | 0.716 | −2.35 ± 0.9 | 519 | - | - |
| GNP-MDS-PLU | 122.96 ±13.6 | 0.482 | −0.99 ± 0.2 | 527 | 84.7 | 27.12 |
| GNP-PVP | 73.56 ±6.6 | 0.603 | −5.07 ± 0.6 | 521 | - | - |
| GNP-MDS-PVP | 231.67 ±23 | 0.247 | −5.29 ± 1.6 | 533 | 93.6 | 30.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatar, A.-S.; Nagy-Simon, T.; Tigu, A.B.; Tomuleasa, C.; Boca, S. Optimization of Tyrosine Kinase Inhibitor-Loaded Gold Nanoparticles for Stimuli-Triggered Antileukemic Drug Release. J. Funct. Biomater. 2023, 14, 399. https://doi.org/10.3390/jfb14080399
Tatar A-S, Nagy-Simon T, Tigu AB, Tomuleasa C, Boca S. Optimization of Tyrosine Kinase Inhibitor-Loaded Gold Nanoparticles for Stimuli-Triggered Antileukemic Drug Release. Journal of Functional Biomaterials. 2023; 14(8):399. https://doi.org/10.3390/jfb14080399
Chicago/Turabian StyleTatar, Andra-Sorina, Timea Nagy-Simon, Adrian Bogdan Tigu, Ciprian Tomuleasa, and Sanda Boca. 2023. "Optimization of Tyrosine Kinase Inhibitor-Loaded Gold Nanoparticles for Stimuli-Triggered Antileukemic Drug Release" Journal of Functional Biomaterials 14, no. 8: 399. https://doi.org/10.3390/jfb14080399