
Engineering Degradation Rate of Polyphosphazene-Based Layer-by-Layer Polymer Coatings

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Dynamic Light Scattering (DLS)
2.3. Residual Molecular Weight
2.4. Size-Exclusion Chromatography (SEC)
2.5. Asymmetric Flow Field Flow Fractionation (AF4)
2.6. Layer-by-Layer (LbL) Film Deposition
2.7. Spectroscopic Ellipsometry
2.8. Quartz Crystal Microbalance (QCM) Measurements
2.9. Fourier Transform Infrared (FTIR) Spectroscopy
2.10. Optical and Scanning Electron Microscopy
3. Results
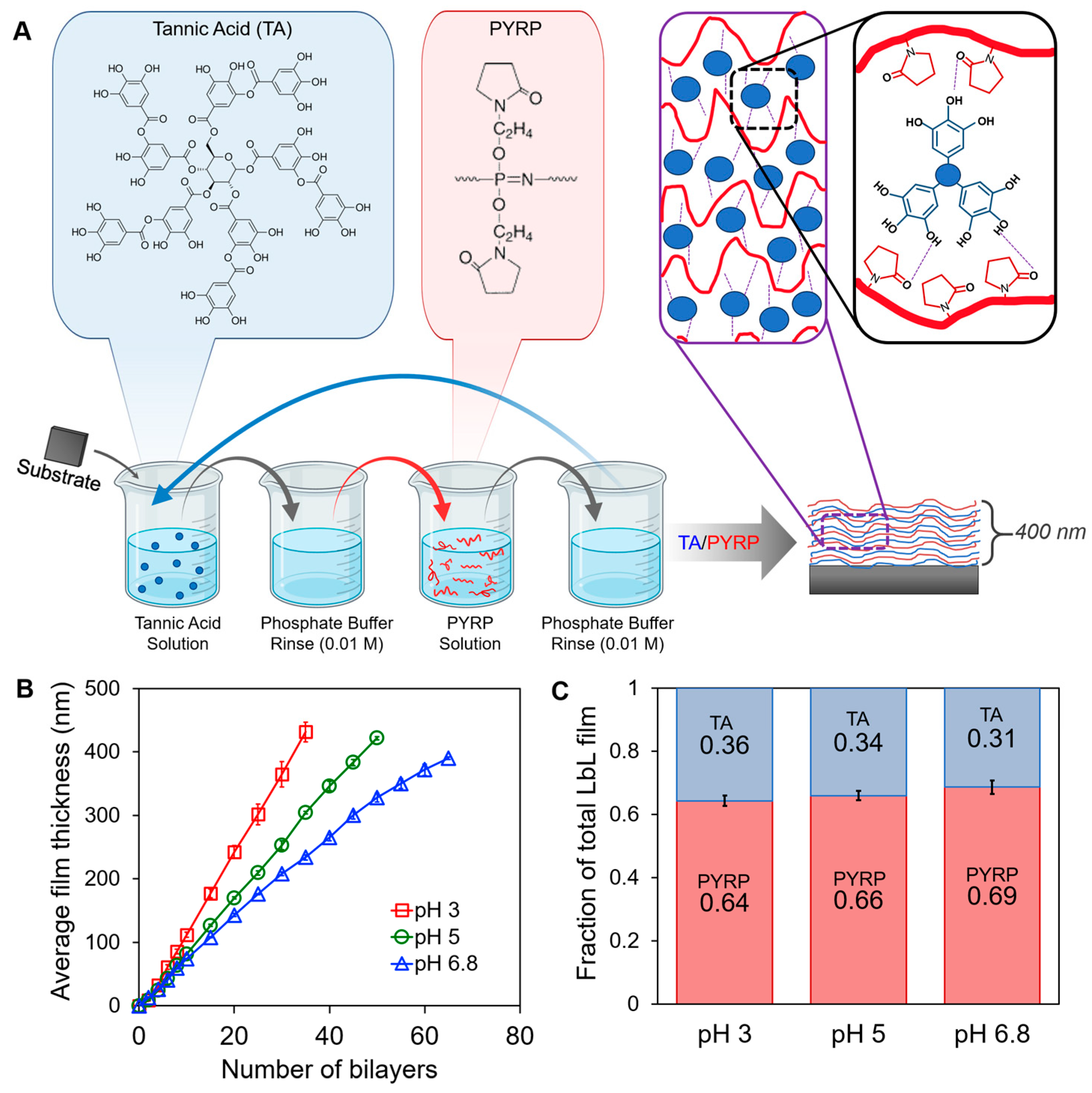
3.1. Layer-by-Layer Assembly of PYRP/TA at pH 3, 5, and 6.8
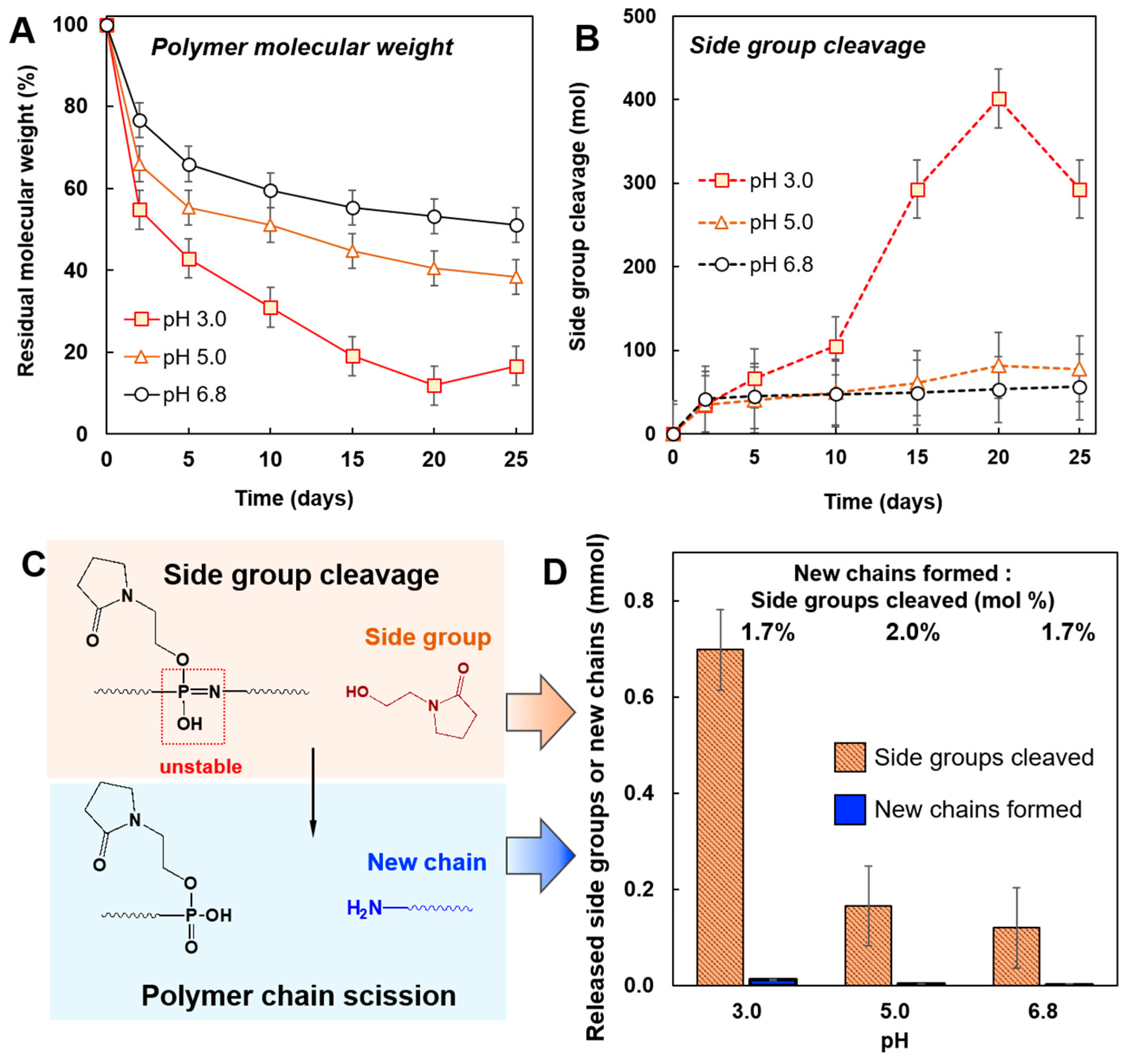
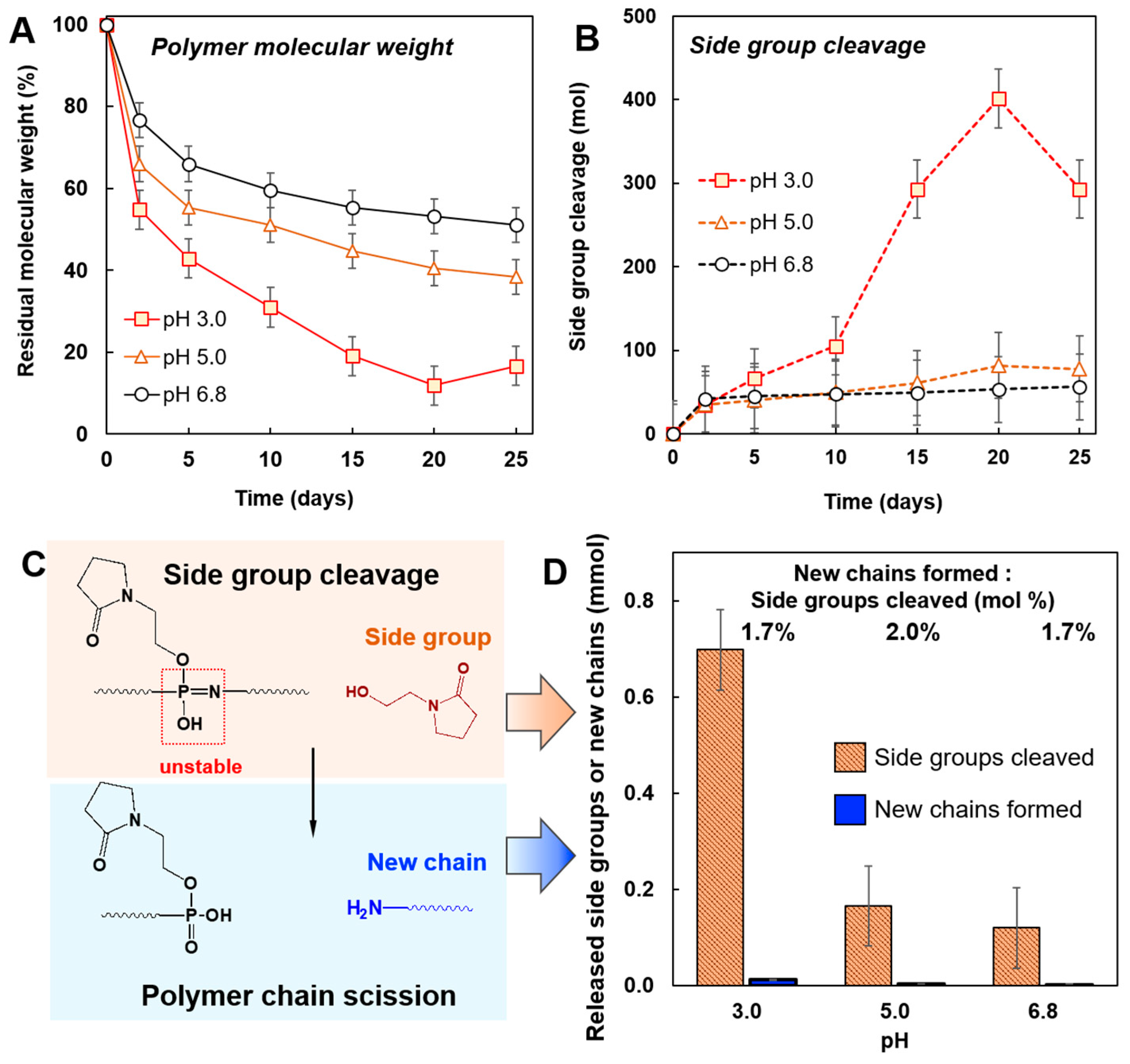
3.2. Degradation of PYRP
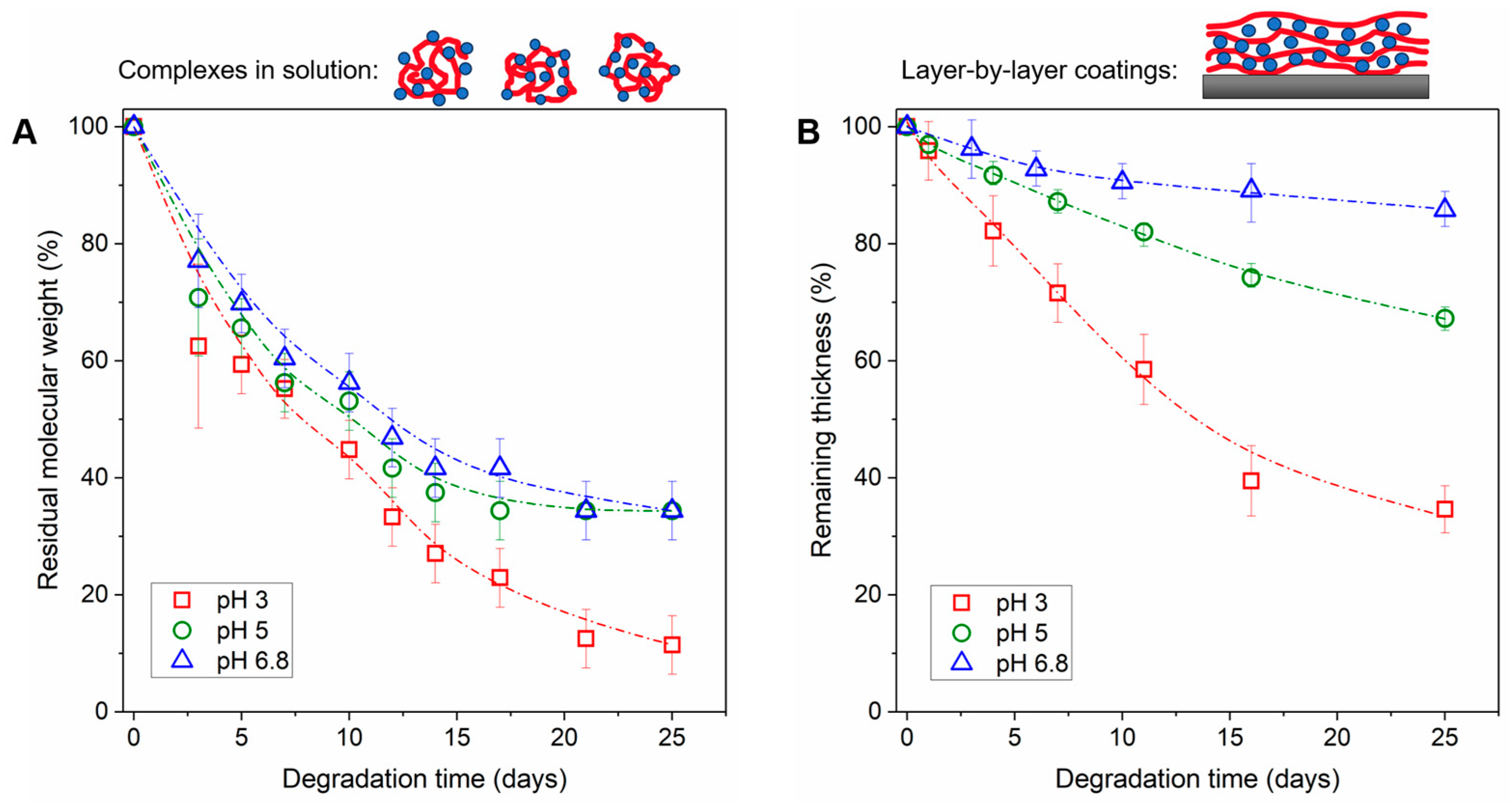
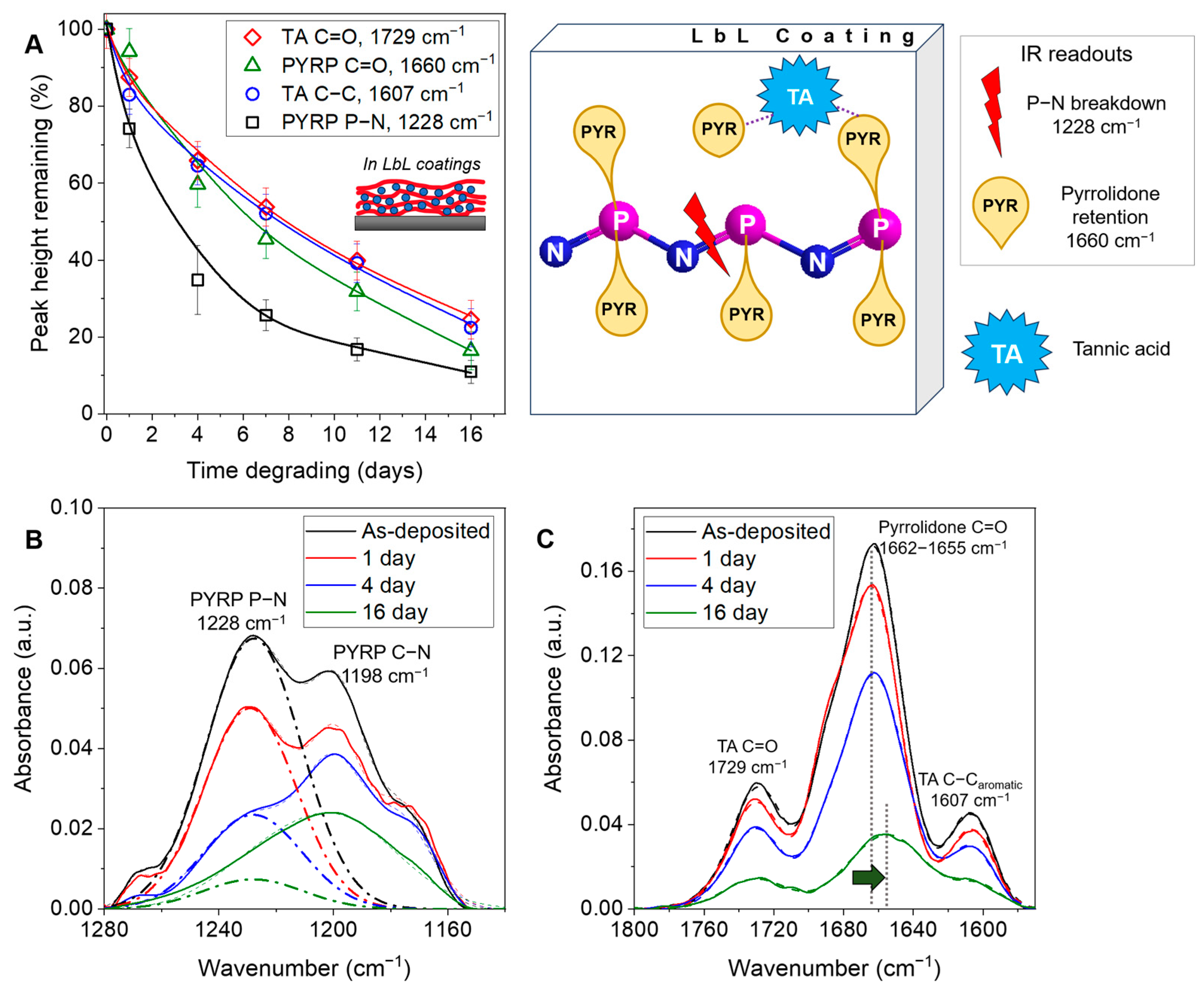
3.3. Degradation of PYRP/TA Complexes in Solution and LbL Coatings
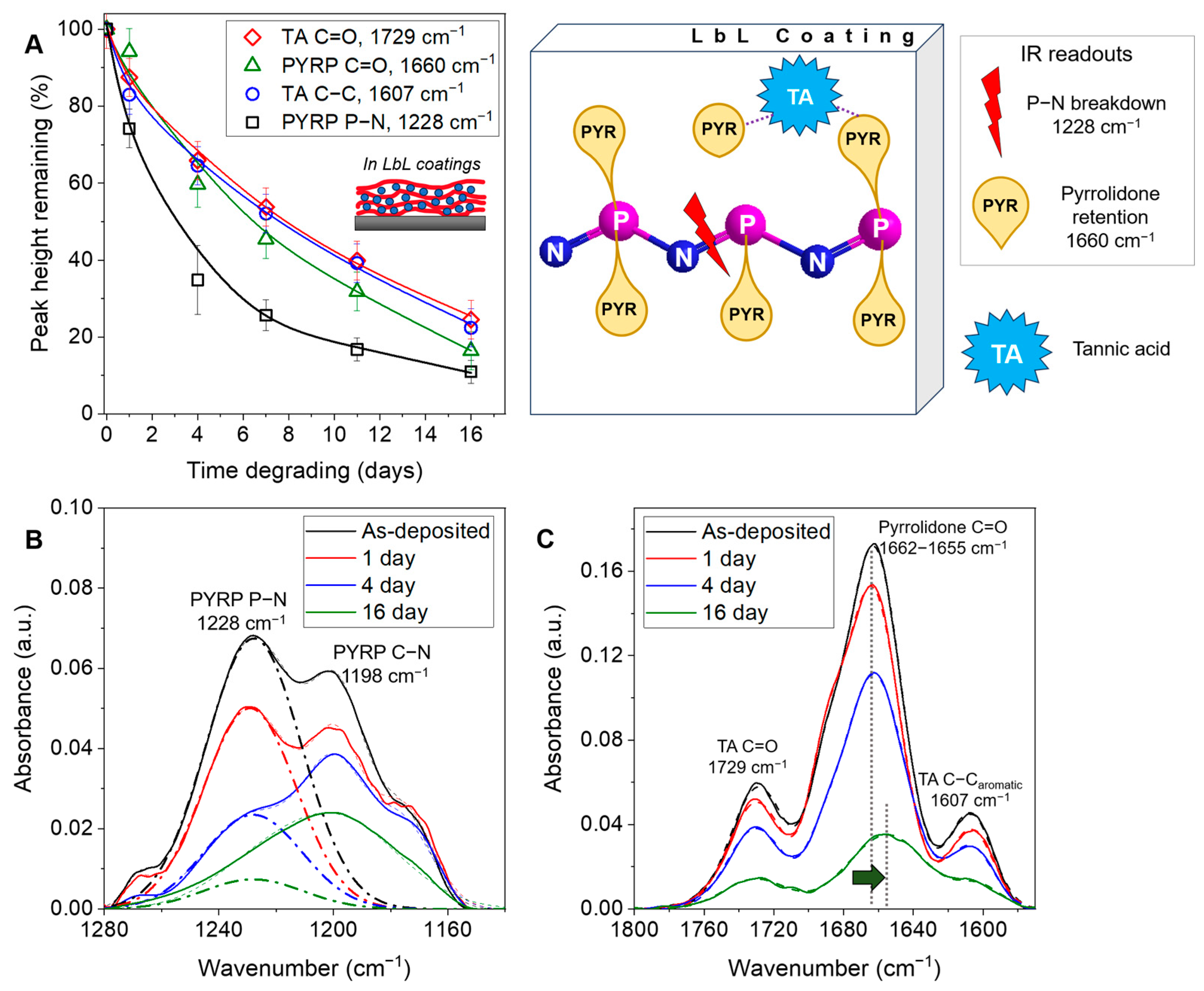
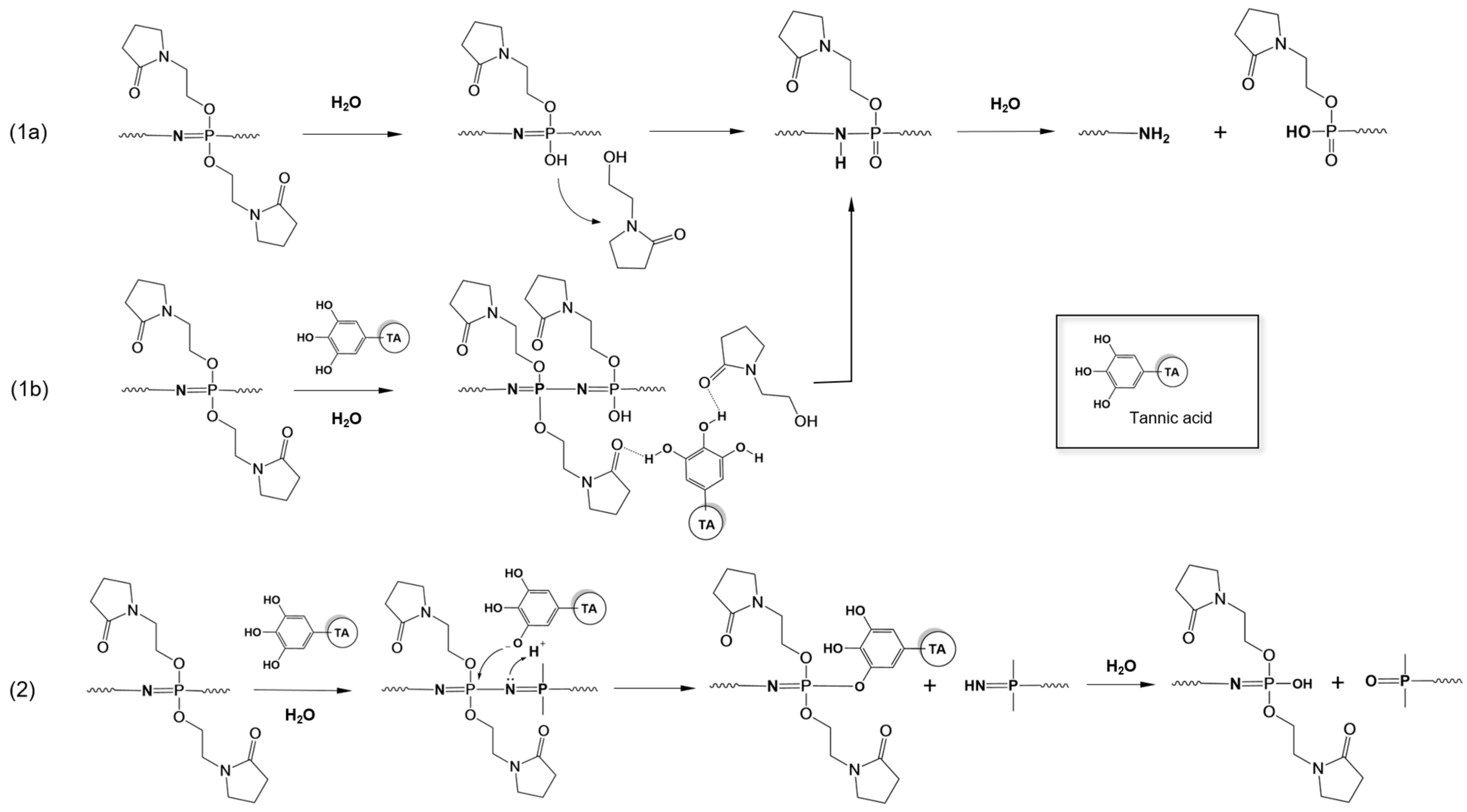
3.4. Molecular Mechanism of LbL Coating Degradation
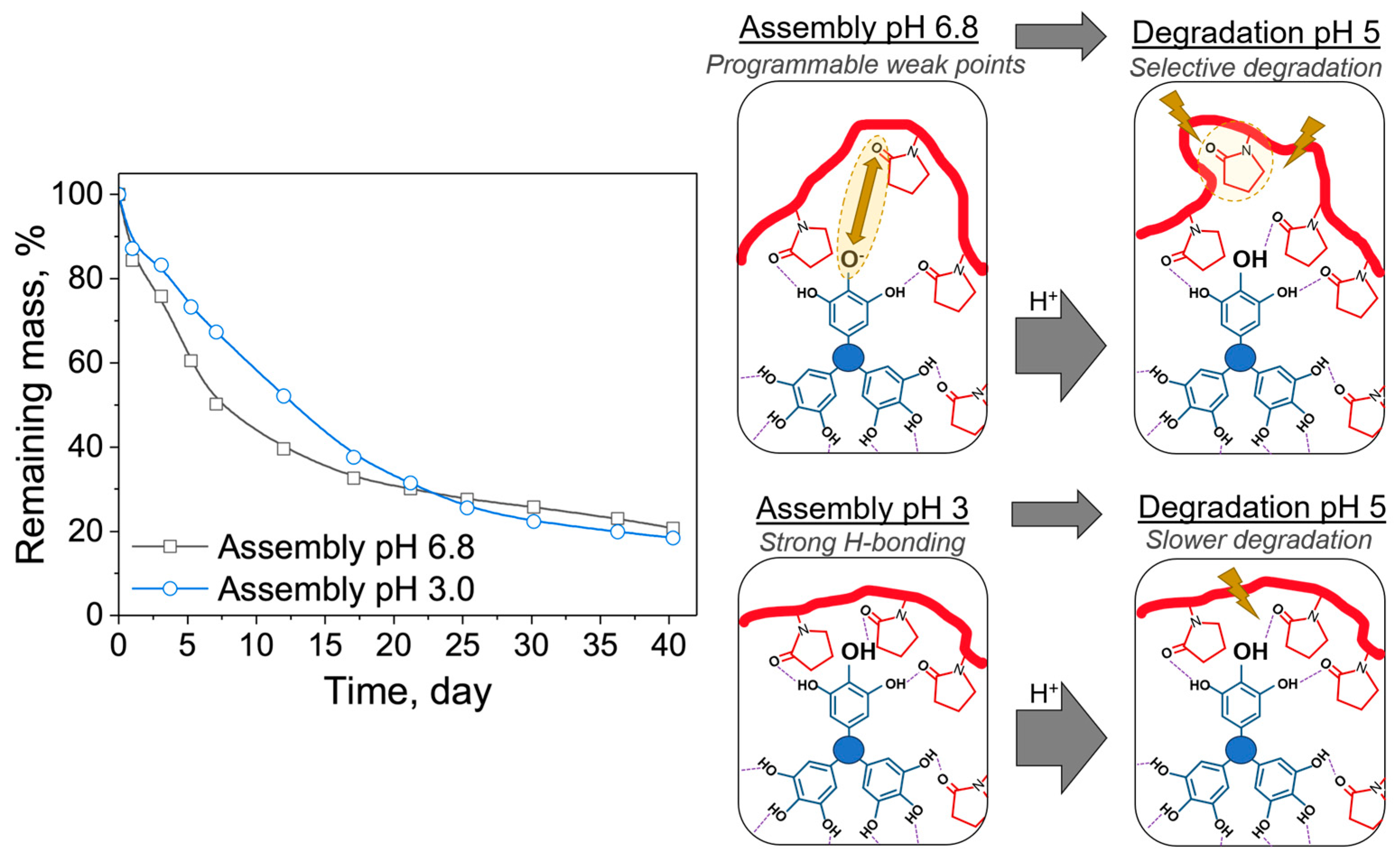
3.5. Effect of Initial Assembly pH on Degradation: Assembly pH 3 and 6.8 versus Degradation at pH 5
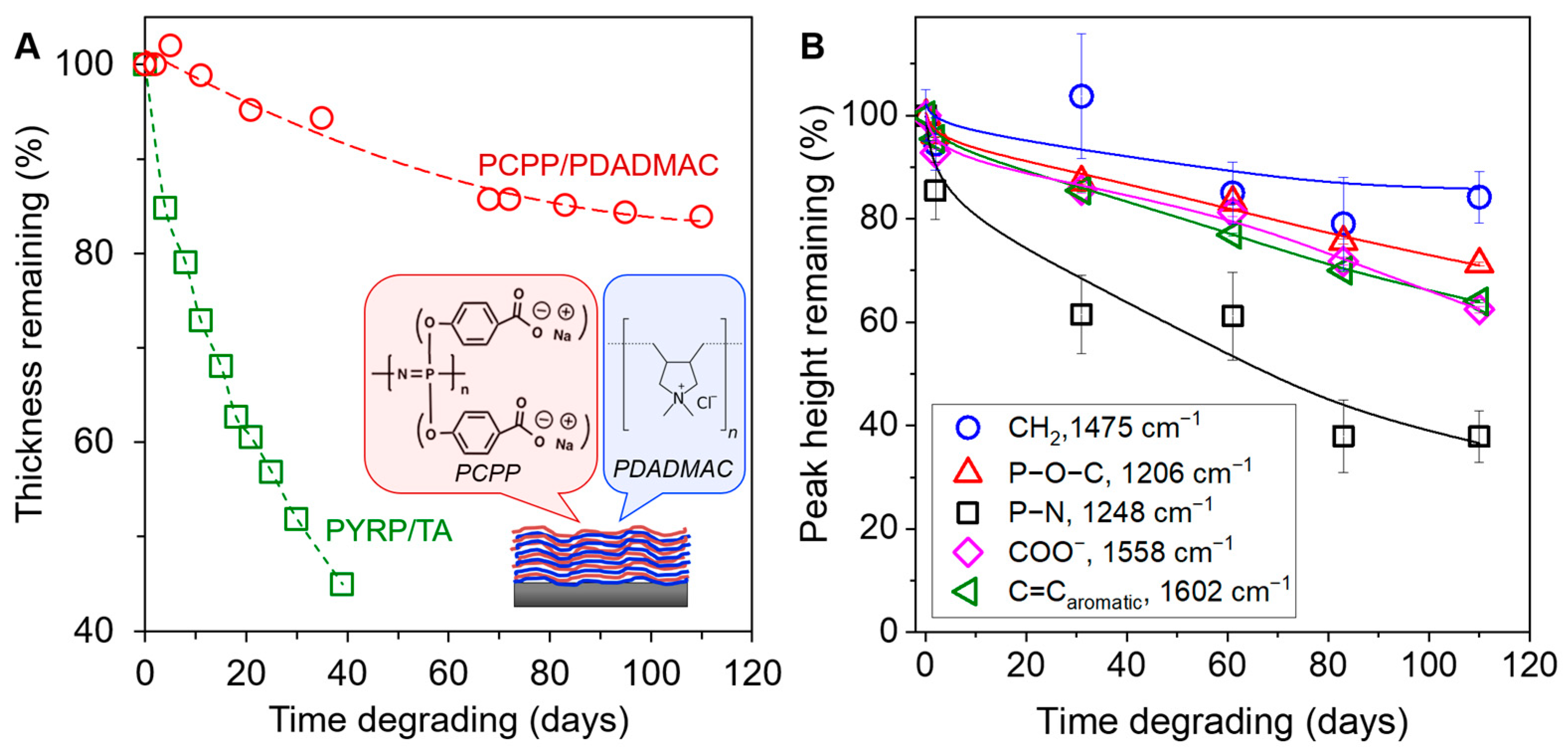
3.6. Effect of Polyphosphazene Side Group on the Degradation of LbL Coatings: PYRP/TA versus PCPP/PDADMAC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Calori, I.R.; Braga, G.; de Jesus, P.d.C.C.; Bi, H.; Tedesco, A.C. Polymer scaffolds as drug delivery systems. Eur. Polym. J. 2020, 129, 109621. [Google Scholar] [CrossRef]
- Adepu, S.; Ramakrishna, S. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.K.; Kim, S.W. Recent advances in polymeric drug delivery systems. Biomater. Res. 2020, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Alkekhia, D.; Hammond, P.T.; Shukla, A. Layer-by-Layer Biomaterials for Drug Delivery. Annu. Rev. Biomed. Eng. 2020, 22, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, R.; Groth, T.W.; Raichur, A.M. Recent Developments in Layer-by-Layer Technique for Drug Delivery Applications. ACS Appl. Bio Mater. 2019, 2, 5512–5527. [Google Scholar] [CrossRef]
- Kim, B.-S.; Park, S.W.; Hammond, P.T. Hydrogen-Bonding Layer-by-Layer-Assembled Biodegradable Polymeric Micelles as Drug Delivery Vehicles from Surfaces. ACS Nano 2008, 2, 386–392. [Google Scholar] [CrossRef]
- Wohl, B.M.; Engbersen, J.F.J. Responsive layer-by-layer materials for drug delivery. J. Control. Release 2012, 158, 2–14. [Google Scholar] [CrossRef]
- Ariga, K.; Lvov, Y.M.; Kawakami, K.; Ji, Q.; Hill, J.P. Layer-by-layer self-assembled shells for drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 762–771. [Google Scholar] [CrossRef]
- Ariga, K.; McShane, M.; Lvov, Y.M.; Ji, Q.; Hill, J.P. Layer-by-layer assembly for drug delivery and related applications. Expert Opin. Drug Deliv. 2011, 8, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Ochs, C.J.; Such, G.K.; Caruso, F. Modular Assembly of Layer-by-Layer Capsules with Tailored Degradation Profiles. Langmuir 2011, 27, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Kharlampieva, E.; Sukhishvili, S.A. Hydrogen-Bonded Layer-by-Layer Polymer Films. J. Macromol. Sci. Part C 2006, 46, 377–395. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Zhang, X.; Xu, J. The influence of pH on a hydrogen-bonded assembly film. Soft Matter 2007, 3, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Caruso, F. Facile Tailoring of Film Morphology and Release Properties Using Layer-by-Layer Assembly of Thermoresponsive Materials. Langmuir 2004, 20, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Shutava, T.; Prouty, M.; Kommireddy, D.; Lvov, Y. pH Responsive Decomposable Layer-by-Layer Nanofilms and Capsules on the Basis of Tannic Acid. Macromolecules 2005, 38, 2850–2858. [Google Scholar] [CrossRef]
- Smith, R.C.; Riollano, M.; Leung, A.; Hammond, P.T. Layer-by-Layer Platform Technology for Small-Molecule Delivery. Angew. Chem. Int. Ed. 2009, 48, 8974–8977. [Google Scholar] [CrossRef]
- Becker, A.L.; Johnston, A.P.R.; Caruso, F. Layer-By-Layer-Assembled Capsules and Films for Therapeutic Delivery. Small 2010, 6, 1836–1852. [Google Scholar] [CrossRef]
- Min, J.; Braatz, R.D.; Hammond, P.T. Tunable staged release of therapeutics from layer-by-layer coatings with clay interlayer barrier. Biomaterials 2014, 35, 2507–2517. [Google Scholar] [CrossRef]
- Vázquez, E.; Dewitt, D.M.; Hammond, P.T.; Lynn, D.M. Construction of Hydrolytically-Degradable Thin Films via Layer-by-Layer Deposition of Degradable Polyelectrolytes. J. Am. Chem. Soc. 2002, 124, 13992–13993. [Google Scholar] [CrossRef]
- Moskowitz, J.S.; Blaisse, M.R.; Samuel, R.E.; Hsu, H.-P.; Harris, M.B.; Martin, S.D.; Lee, J.C.; Spector, M.; Hammond, P.T. The effectiveness of the controlled release of gentamicin from polyelectrolyte multilayers in the treatment of Staphylococcus aureus infection in a rabbit bone model. Biomaterials 2010, 31, 6019–6030. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Avadhany, S.N.; Fang, J.C.; Hammond, P.T. Tunable Vancomycin Releasing Surfaces for Biomedical Applications. Small 2010, 6, 2392–2404. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.M.; Langer, R. Degradable Poly(β-amino esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA. J. Am. Chem. Soc. 2000, 122, 10761–10768. [Google Scholar] [CrossRef]
- Chou, J.J.; Berger, A.G.; Jalili-Firoozinezhad, S.; Hammond, P.T. A design approach for layer-by-layer surface-mediated siRNA delivery. Acta Biomater. 2021, 135, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Appadoo, V.; Carter, M.C.D.; Lynn, D.M. Controlling the surface-mediated release of DNA using ‘mixed multilayers’. Bioeng. Transl. Med. 2016, 1, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.F.; Smith, R.C.; Hammond, P.T. Polyelectrolyte Multilayers for Tunable Release of Antibiotics. Biomacromolecules 2008, 9, 1660–1668. [Google Scholar] [CrossRef]
- Macdonald, M.; Rodriguez, N.M.; Smith, R.; Hammond, P.T. Release of a model protein from biodegradable self assembled films for surface delivery applications. J. Control. Release 2008, 131, 228–234. [Google Scholar] [CrossRef]
- Wood, K.C.; Boedicker, J.Q.; Lynn, D.M.; Hammond, P.T. Tunable Drug Release from Hydrolytically Degradable Layer-by-Layer Thin Films. Langmuir 2005, 21, 1603–1609. [Google Scholar] [CrossRef]
- Wei, J.; Zhu, L.; Lu, Q.; Li, G.; Zhou, Y.; Yang, Y.; Zhang, L. Recent progress and applications of poly(beta amino esters)-based biomaterials. J. Control. Release 2023, 354, 337–353. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Keskin, D.; Shi, L. Poly(β-Amino Esters): Synthesis, Formulations, and Their Biomedical Applications. Adv. Healthc. Mater. 2018, 8, 1801359. [Google Scholar] [CrossRef]
- Chen, F.; Teniola, O.R.; Laurencin, C.T. Biodegradable polyphosphazenes for regenerative engineering. J. Mater. Res. 2022, 37, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(Amino Acid Ester)Phosphazenes] as Substrates for the Controlled-Release of Small Molecules. Biomaterials 1994, 15, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Ogueri, K.S.; Ogueri, K.S.; Allcock, H.R.; Laurencin, C.T. Polyphosphazene polymers: The next generation of biomaterials for regenerative engineering and therapeutic drug delivery. J. Vac. Sci. Technol. B 2020, 38, 030801. [Google Scholar] [CrossRef] [PubMed]
- Tagad, H.D.; Brito, J.; Marin, A.; Buckley, C.; Wang, H.; Sun, J.; Sukhishvili, S.A.; Wang, H.; Andrianov, A.K. 4-Methylumbelliferone-Functionalized Polyphosphazene and Its Assembly into Biocompatible Fluorinated Nanocoatings with Selective Antiproliferative Activity. Biomacromolecules 2023, 24, 2278–2290. [Google Scholar] [CrossRef] [PubMed]
- Chun, C.; Lee, S.M.; Kim, S.Y.; Yang, H.K.; Song, S.-C. Thermosensitive poly(organophosphazene)–paclitaxel conjugate gels for antitumor applications. Biomaterials 2009, 30, 2349–2360. [Google Scholar] [CrossRef]
- Cho, J.-K.; Chun, C.; Kuh, H.-J.; Song, S.-C. Injectable poly(organophosphazene)–camptothecin conjugate hydrogels: Synthesis, characterization, and antitumor activities. Eur. J. Pharm. Biopharm. 2012, 81, 582–590. [Google Scholar] [CrossRef]
- Teasdale, I.; Waser, M.; Wilfert, S.; Falk, H.; Brüggemann, O. Photoreactive, water-soluble conjugates of hypericin with polyphosphazenes. Monatshefte Chem.-Chem. Mon. 2012, 143, 355–360. [Google Scholar] [CrossRef]
- Teasdale, I.; Wilfert, S.; Nischang, I.; Brüggemann, O. Multifunctional and biodegradable polyphosphazenes for use as macromolecular anti-cancer drug carriers. Polym. Chem. 2011, 2, 828–834. [Google Scholar] [CrossRef]
- Albright, V.; Penarete-Acosta, D.; Stack, M.; Zheng, J.; Marin, A.; Hlushko, H.; Wang, H.; Jayaraman, A.; Andrianov, A.K.; Sukhishvili, S.A. Polyphosphazenes enable durable, hemocompatible, highly efficient antibacterial coatings. Biomaterials 2021, 268, 120586. [Google Scholar] [CrossRef]
- Marin, A.; Brito, J.; Sukhishvili, S.A.; Andrianov, A.K. Cationic Fluoropolyphosphazenes: Synthesis and Assembly with Heparin as a Pathway to Hemocompatible Nanocoatings. ACS Appl. Bio Mater. 2022, 5, 313–321. [Google Scholar] [CrossRef]
- Allcock, H.R.; Morozowich, N.L. Bioerodible polyphosphazenes and their medical potential. Polym. Chem. 2012, 3, 578–590. [Google Scholar] [CrossRef]
- Haudum, S.; Strasser, P.; Teasdale, I. Phosphorus and Silicon-Based Macromolecules as Degradable Biomedical Polymers. Macromol. Biosci. 2023, 2023, 2300127. [Google Scholar] [CrossRef]
- Teasdale, I.; Bruggemann, O. Polyphosphazenes: Multifunctional, Biodegradable Vehicles for Drug and Gene Delivery. Polymer 2013, 5, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Lambert, W.J.; Kudla, R.J.; Holland, J.M.; Curry, J.T. A biodegradable transdermal penetration enhancer based on N-(2-hydroxyethyl)-2-pyrrolidone I. Synthesis and characterization. Int. J. Pharm. 1993, 95, 181–192. [Google Scholar] [CrossRef]
- Kováčik, A.; Kopečná, M.; Vávrová, K. Permeation enhancers in transdermal drug delivery: Benefits and limitations. Expert Opin. Drug Deliv. 2020, 17, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Kaur, M.P. Permeation Enhancers for Transdermal Drug Delivery. Drug Dev. Ind. Pharm. 2000, 26, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Marwah, H.; Garg, T.; Goyal, A.K.; Rath, G. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2016, 23, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Aldawood, F.K.; Andar, A.; Desai, S. A Comprehensive Review of Microneedles: Types, Materials, Processes, Characterizations and Applications. Polymers 2021, 13, 2815. [Google Scholar] [CrossRef]
- Jeong, W.Y.; Kwon, M.; Choi, H.E.; Kim, K.S. Recent advances in transdermal drug delivery systems: A review. Biomater. Res. 2021, 25, 24. [Google Scholar] [CrossRef]
- Brito, J.; Andrianov, A.K.; Sukhishvili, S.A. Factors Controlling Degradation of Biologically Relevant Synthetic Polymers in Solution and Solid State. ACS Appl. Bio Mater. 2022, 5, 5057–5076. [Google Scholar] [CrossRef]
- Baldwin, A.; Booth, B.W. Biomedical applications of tannic acid. J. Biomater. Appl. 2022, 36, 1503–1523. [Google Scholar] [CrossRef] [PubMed]
- Erel-Unal, I.; Sukhishvili, S.A. Hydrogen-Bonded Multilayers of a Neutral Polymer and a Polyphenol. Macromolecules 2008, 41, 3962–3970. [Google Scholar] [CrossRef]
- Andrianov, A.K.; Marin, A.; Peterson, P. Water-soluble biodegradable polyphosphazenes containing N-ethylpyrrolidone groups. Macromolecules 2005, 38, 7972–7976. [Google Scholar] [CrossRef]
- Decollibus, D.P.; Marin, A.; Andrianov, A.K. Effect of environmental factors on hydrolytic degradation of water-soluble polyphosphazene polyelectrolyte in aqueous solutions. Biomacromolecules 2010, 11, 2033–2038. [Google Scholar] [CrossRef]
- Gangwar, C.; Yaseen, B.; Kumar, I.; Singh, N.K.; Naik, R.M. Growth Kinetic Study of Tannic Acid Mediated Monodispersed Silver Nanoparticles Synthesized by Chemical Reduction Method and Its Characterization. ACS Omega 2021, 6, 22344–22356. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, A.K.; Langer, R. Polyphosphazene immunoadjuvants: Historical perspective and recent advances. J. Control. Release 2021, 329, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Magiri, R.; Mutwiri, G.; Wilson, H.L. Recent advances in experimental polyphosphazene adjuvants and their mechanisms of action. Cell Tissue Res. 2018, 374, 465–471. [Google Scholar] [CrossRef]
- Hsu, W.-H.; Csaba, N.; Alexander, C.; Garcia-Fuentes, M. Polyphosphazenes for the delivery of biopharmaceuticals. J. Appl. Polym. Sci. 2020, 137, 48688. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(amino acid ester)phosphazenes]: Synthesis, Crystallinity, and Hydrolytic Sensitivity in Solution and the Solid State. Macromolecules 1994, 27, 1071–1075. [Google Scholar] [CrossRef]
- Addy, P.S.; Shivrayan, M.; Cencer, M.; Zhuang, J.; Moore, J.S.; Thayumanavan, S. Polymer with Competing Depolymerization Pathways: Chain Unzipping versus Chain Scission. ACS Macro Lett. 2020, 9, 855–859. [Google Scholar] [CrossRef]
- Allcock, H.R. Chemistry and Applications of Polyphosphazenes; Wiley: Hoboken, NJ, USA, 2002; p. 725. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brito, J.; Moon, J.; Hlushko, R.; Aliakseyeu, A.; Andrianov, A.K.; Sukhishvili, S.A. Engineering Degradation Rate of Polyphosphazene-Based Layer-by-Layer Polymer Coatings. J. Funct. Biomater. 2024, 15, 26. https://doi.org/10.3390/jfb15020026
Brito J, Moon J, Hlushko R, Aliakseyeu A, Andrianov AK, Sukhishvili SA. Engineering Degradation Rate of Polyphosphazene-Based Layer-by-Layer Polymer Coatings. Journal of Functional Biomaterials. 2024; 15(2):26. https://doi.org/10.3390/jfb15020026
Chicago/Turabian StyleBrito, Jordan, Junho Moon, Raman Hlushko, Aliaksei Aliakseyeu, Alexander K. Andrianov, and Svetlana A. Sukhishvili. 2024. "Engineering Degradation Rate of Polyphosphazene-Based Layer-by-Layer Polymer Coatings" Journal of Functional Biomaterials 15, no. 2: 26. https://doi.org/10.3390/jfb15020026
APA StyleBrito, J., Moon, J., Hlushko, R., Aliakseyeu, A., Andrianov, A. K., & Sukhishvili, S. A. (2024). Engineering Degradation Rate of Polyphosphazene-Based Layer-by-Layer Polymer Coatings. Journal of Functional Biomaterials, 15(2), 26. https://doi.org/10.3390/jfb15020026