Antimicrobial Monomers for Polymeric Dental Restoratives: Cytotoxicity and Physicochemical Properties

Abstract
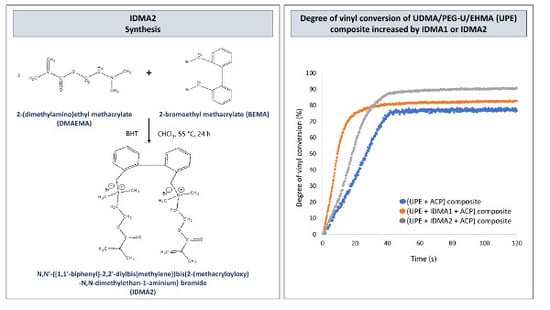
:
1. Introduction
2. Results
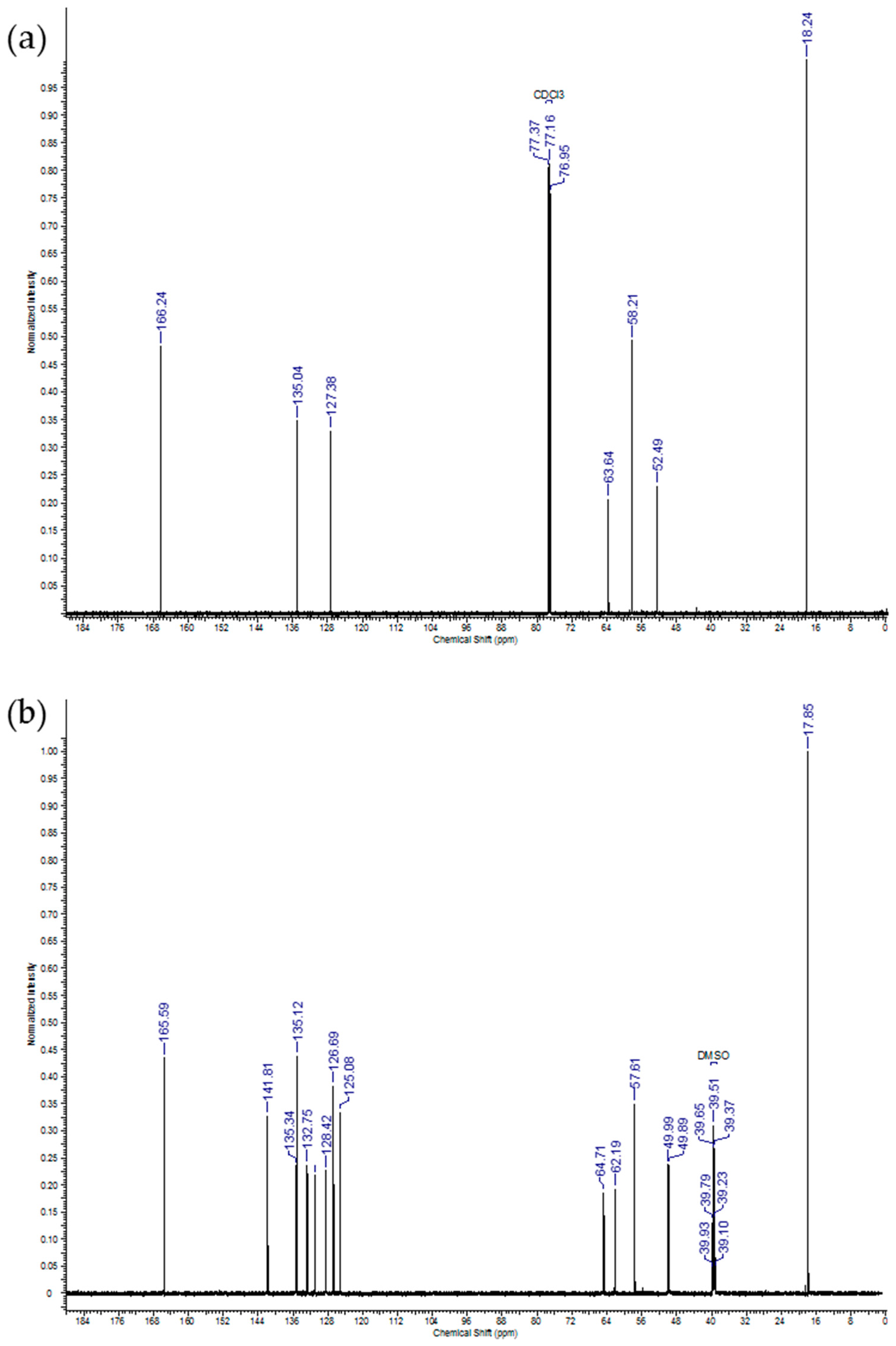
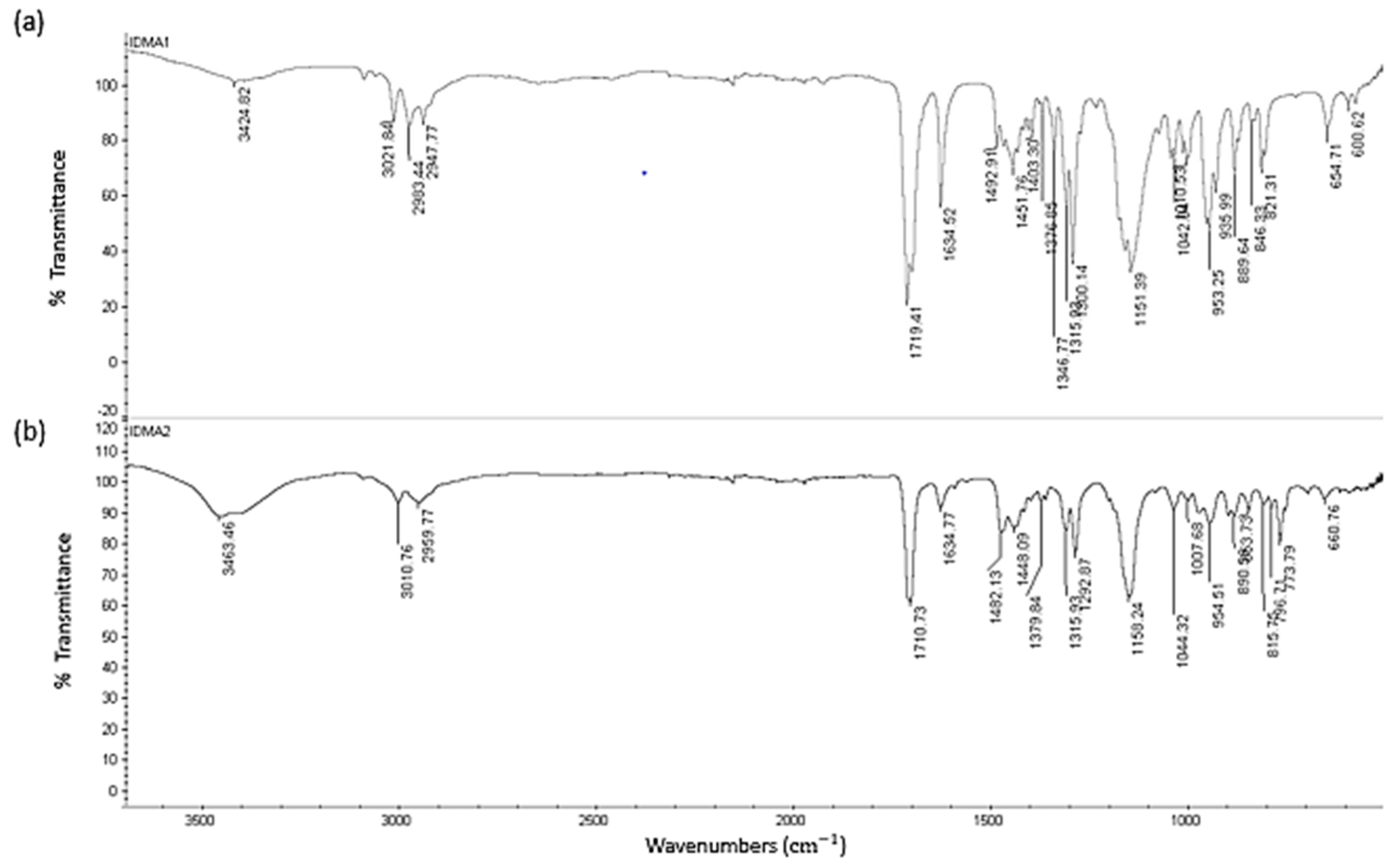
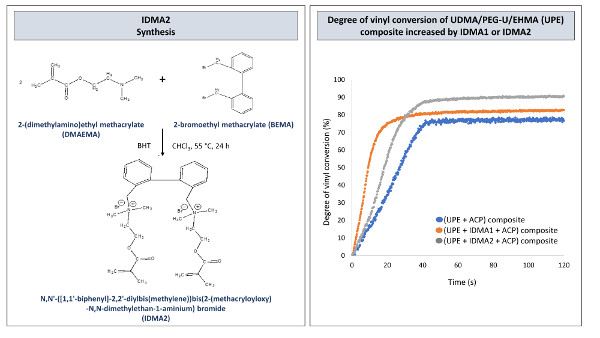
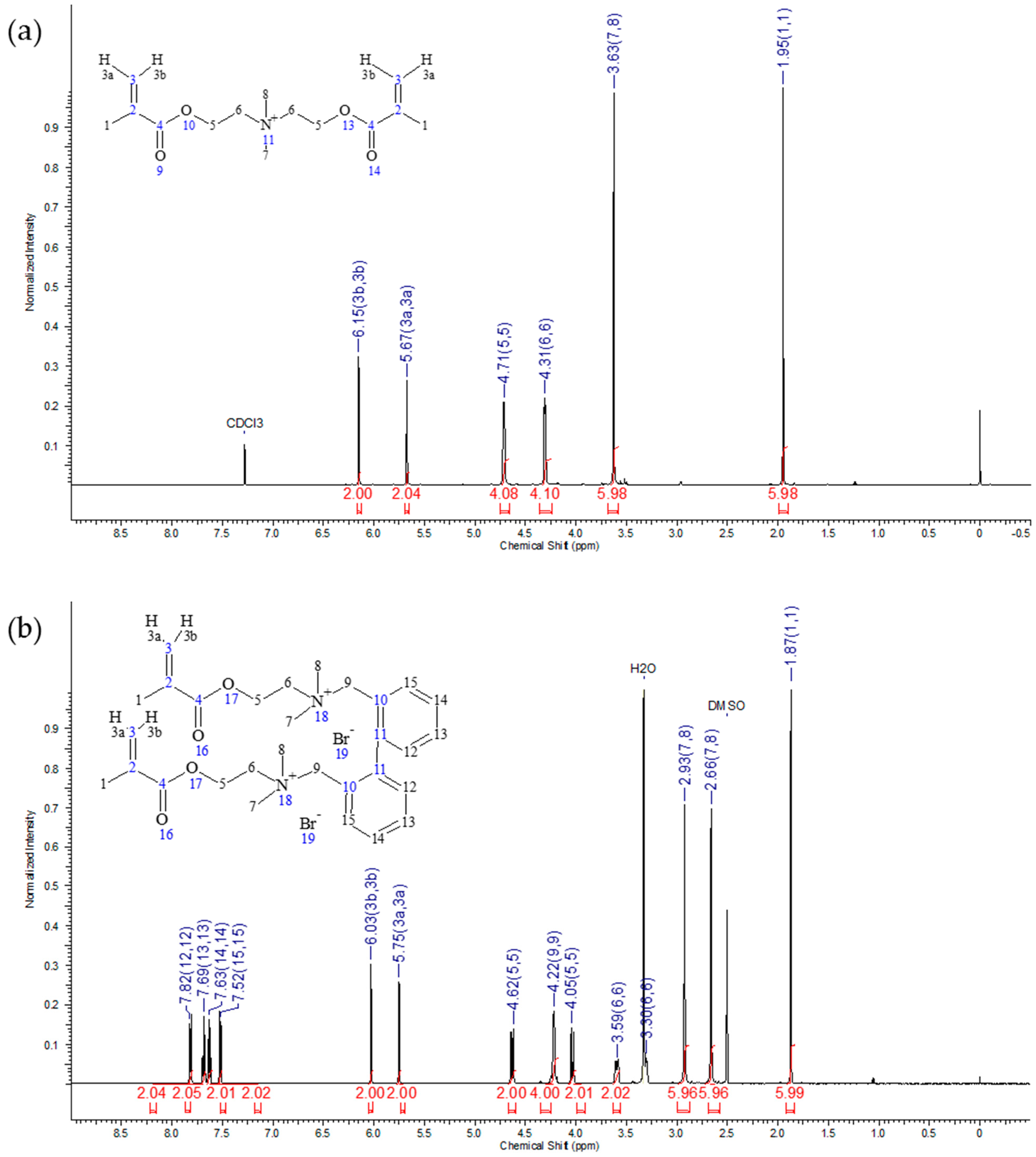
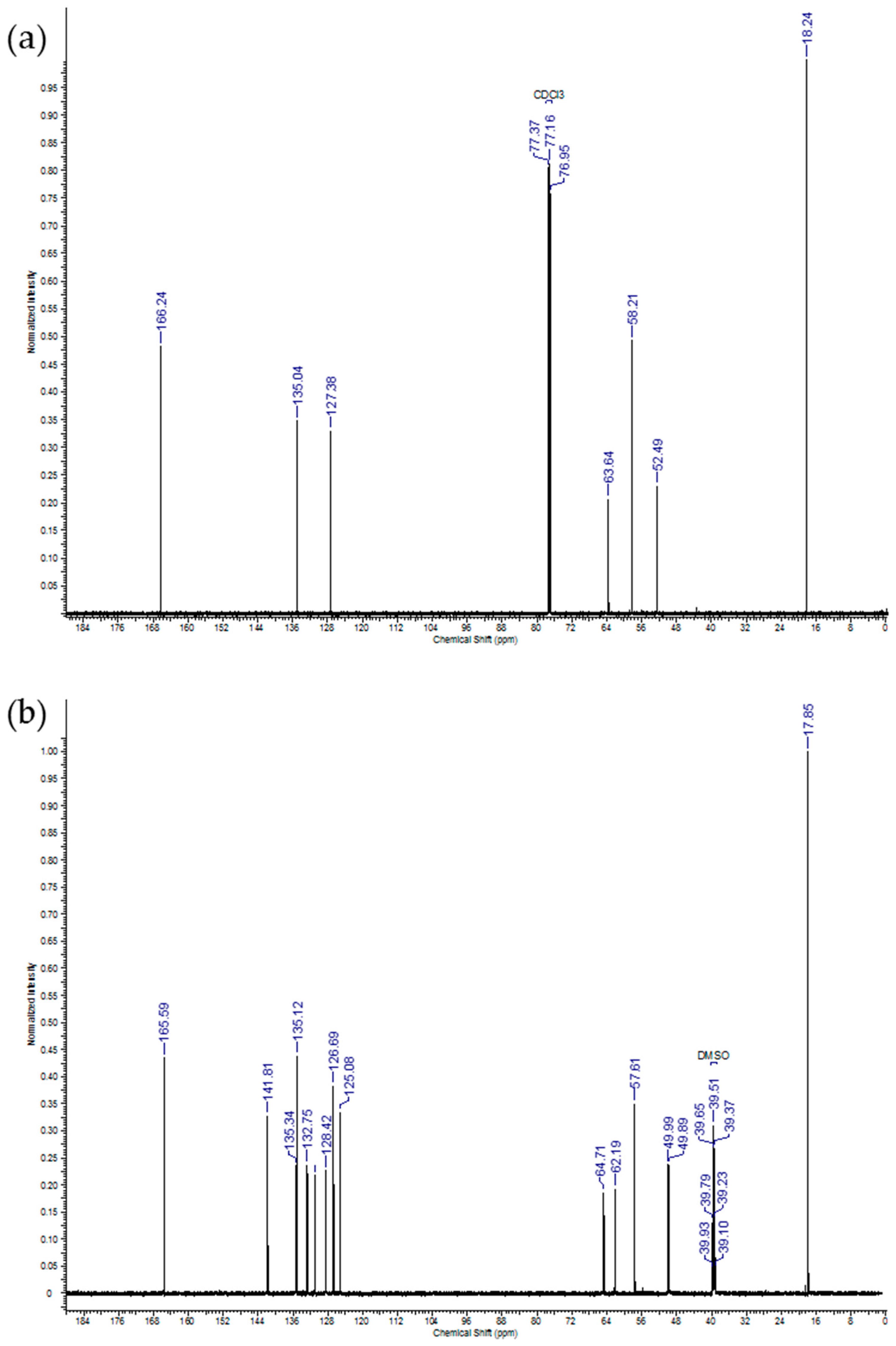
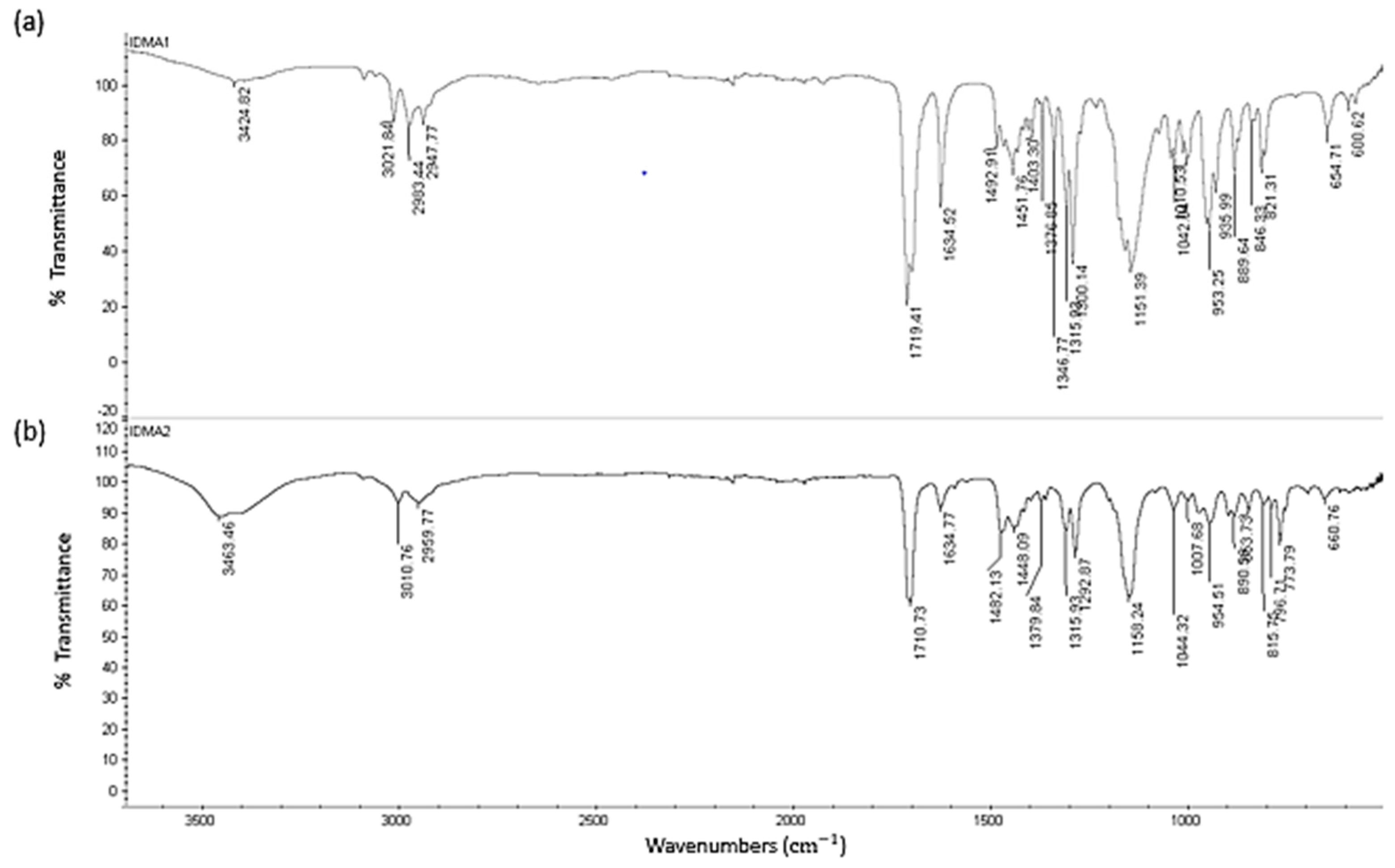
2.1. IDMAs: Structural Validation
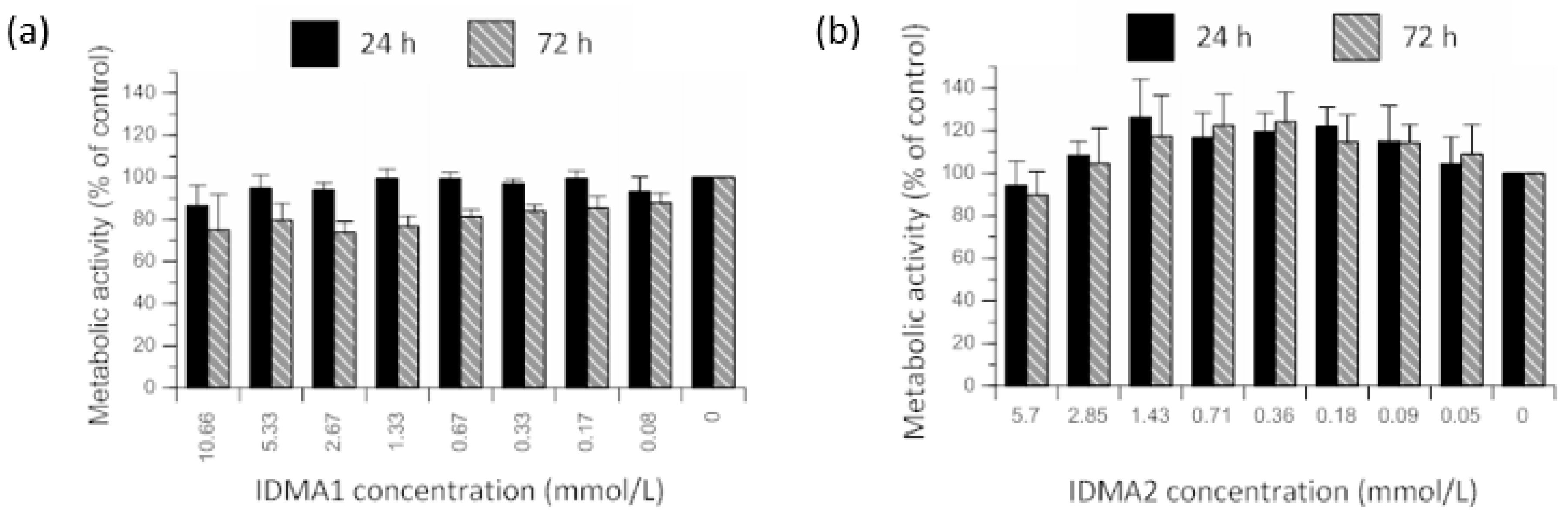
2.2. IDMAs: Biocompatibility
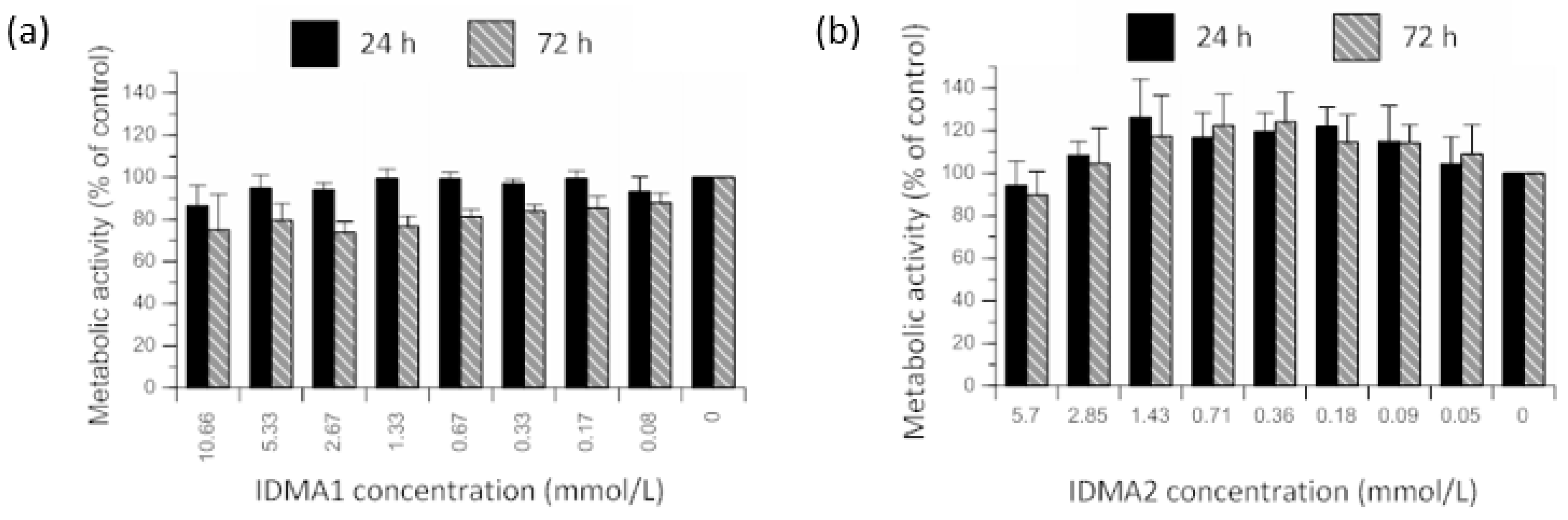
2.2.1. Cellular Metabolic Activity
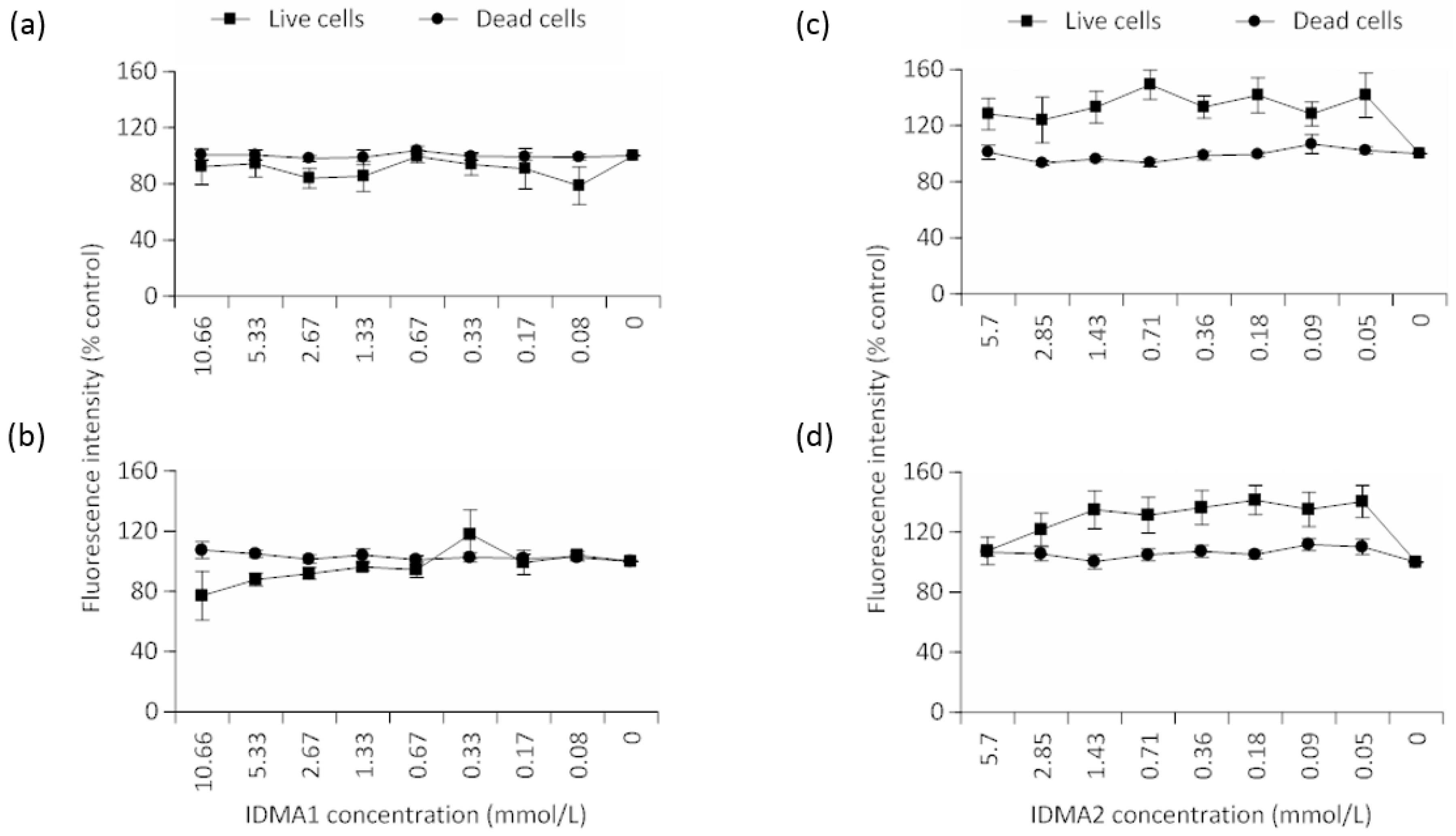
2.2.2. Cellular Viability
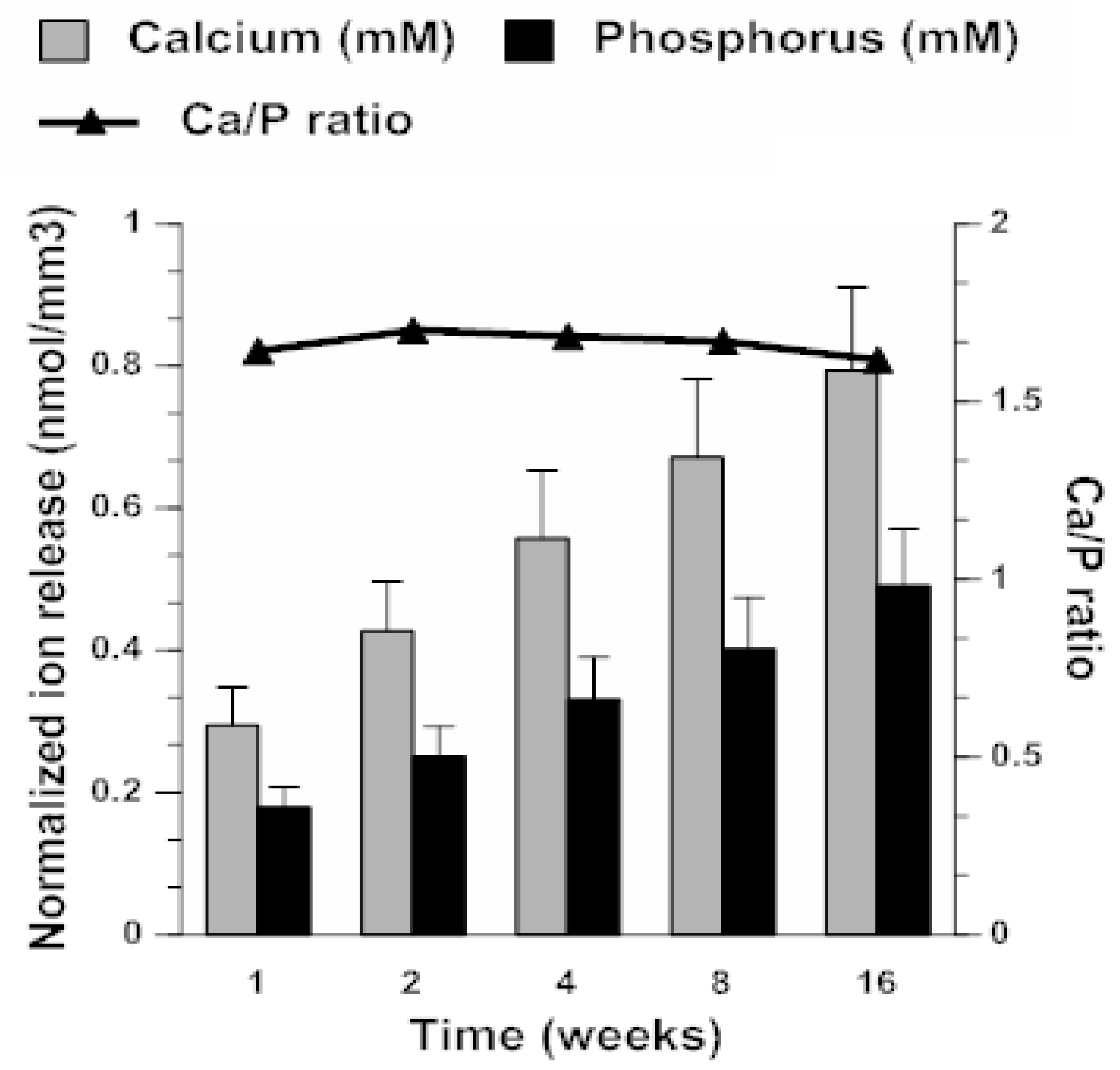
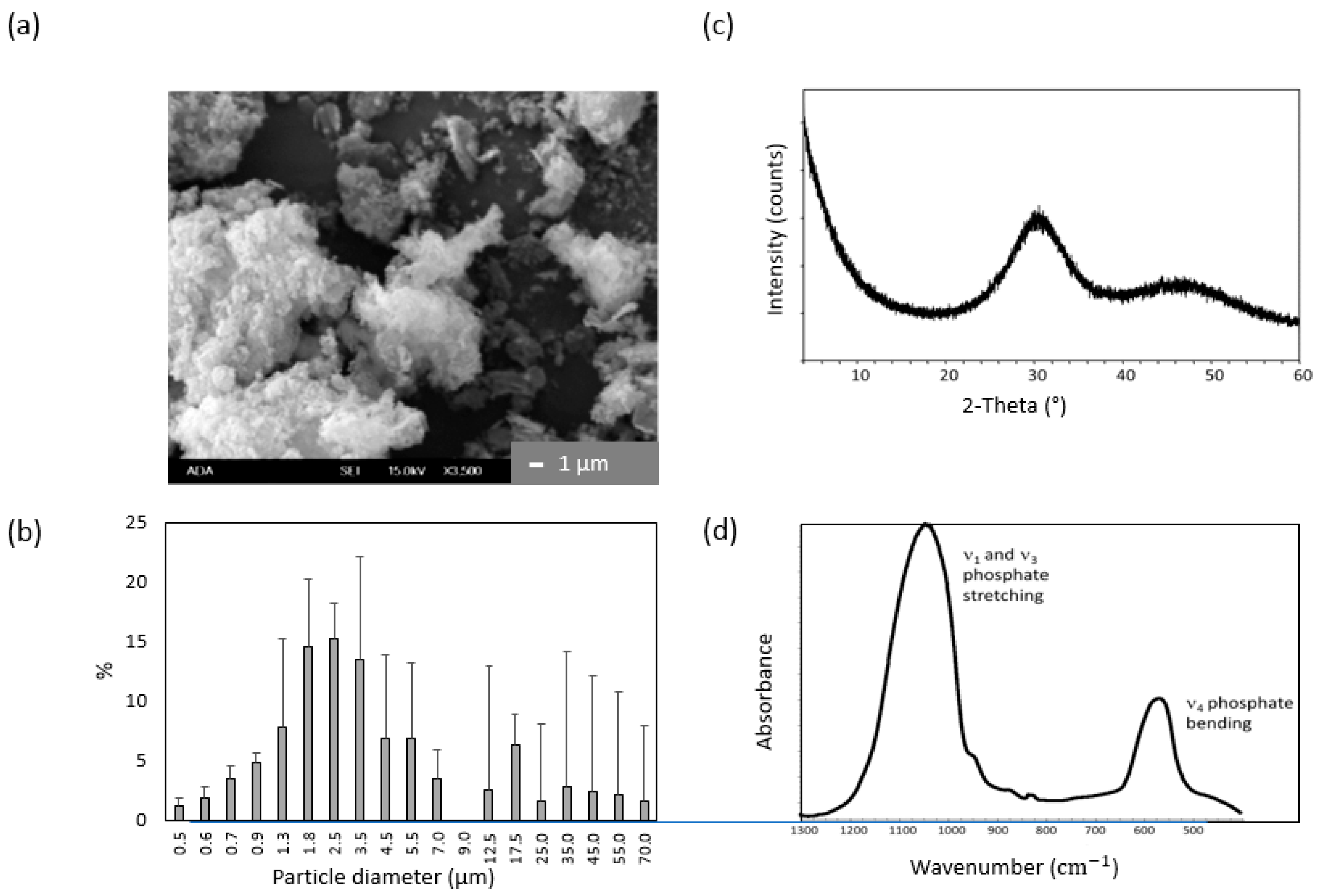
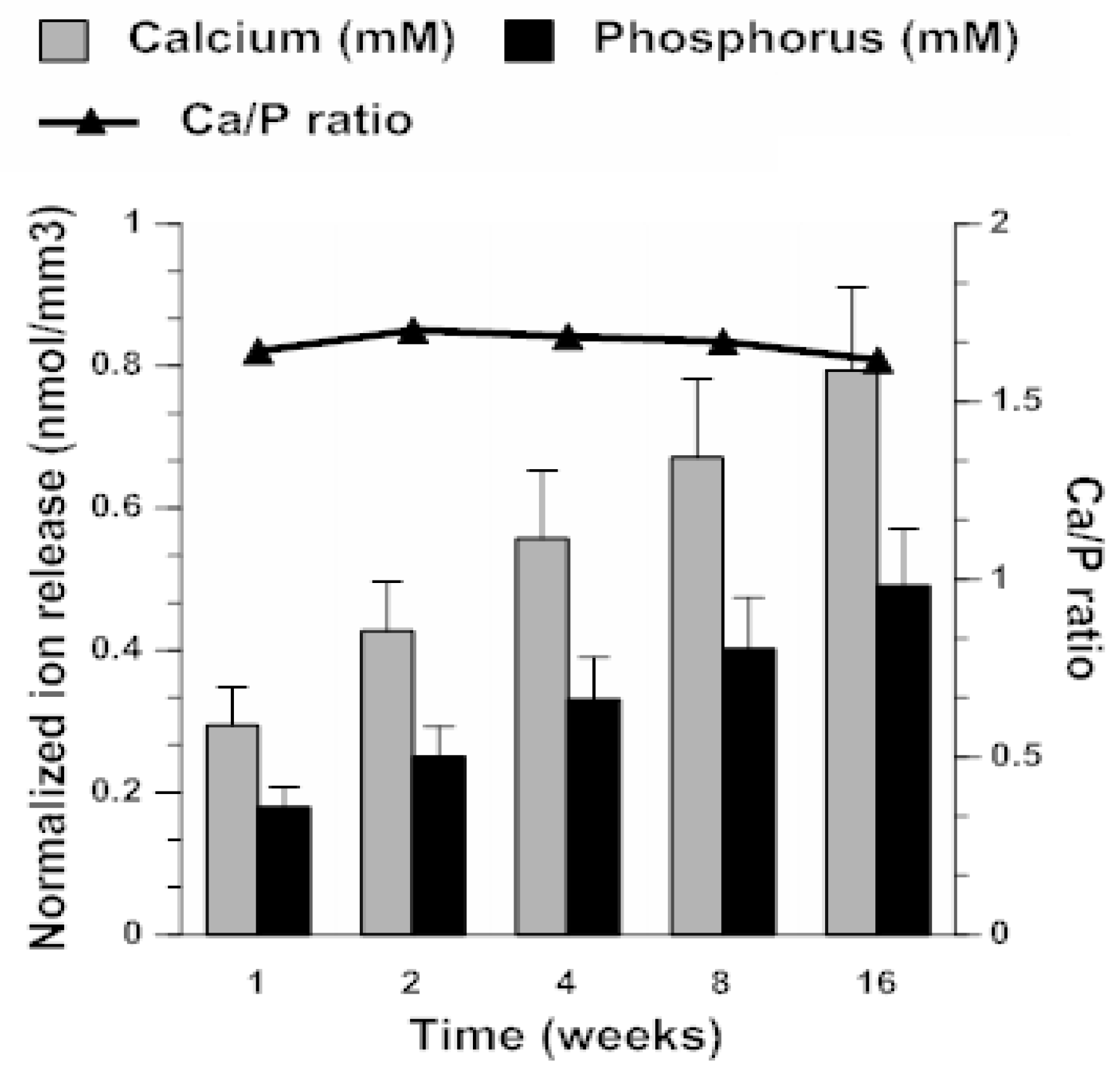
2.3. ACP Filler Remineralizing Ability
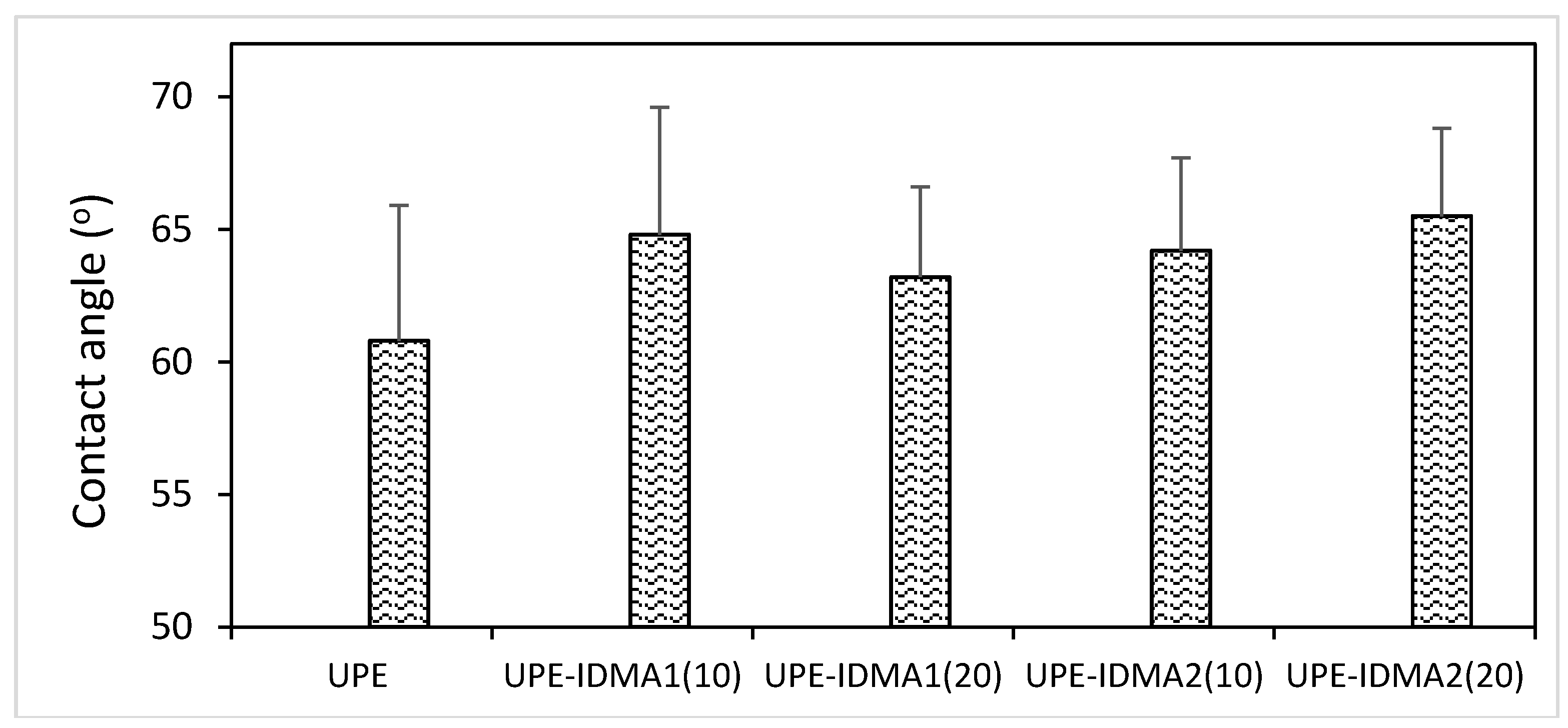
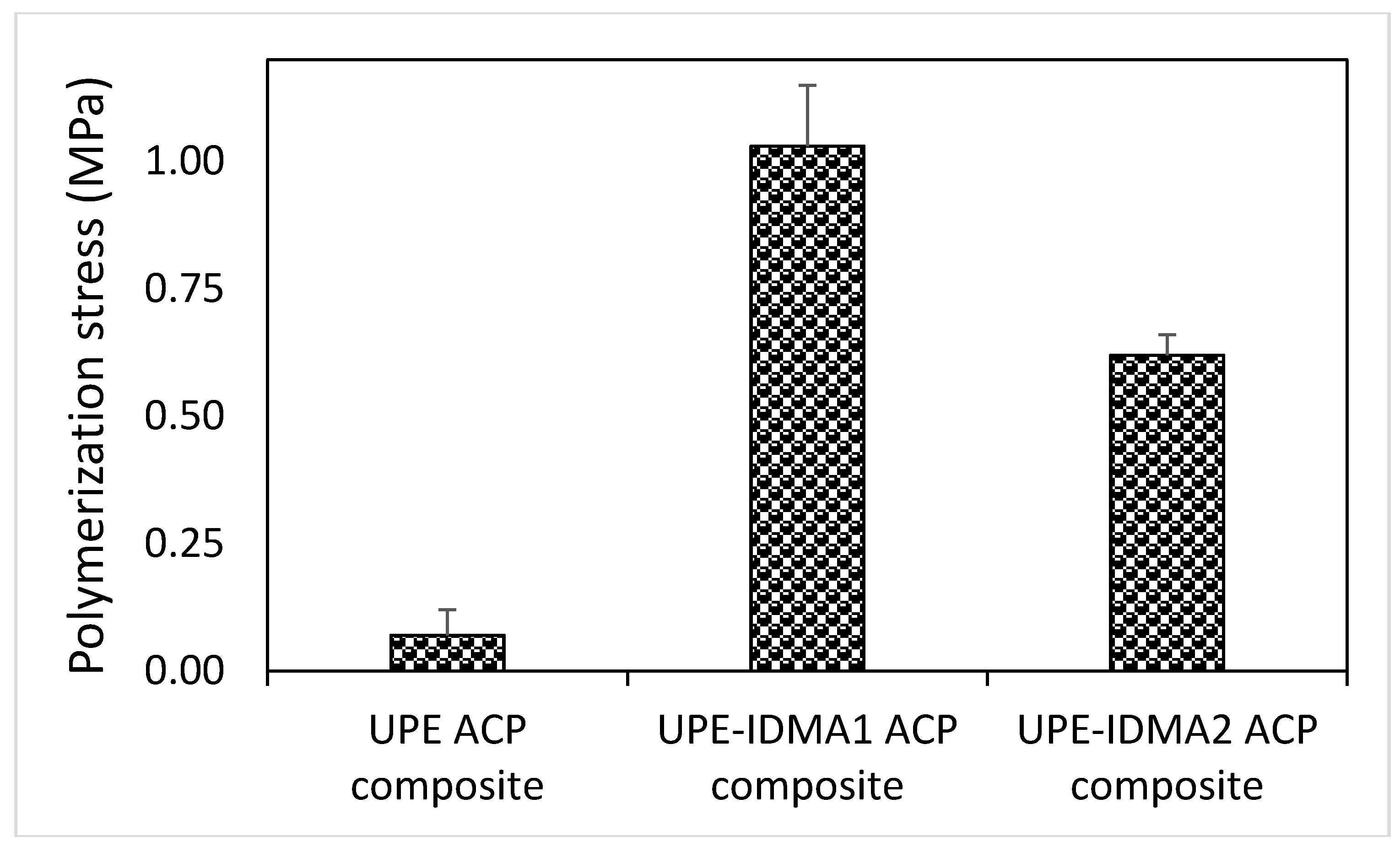
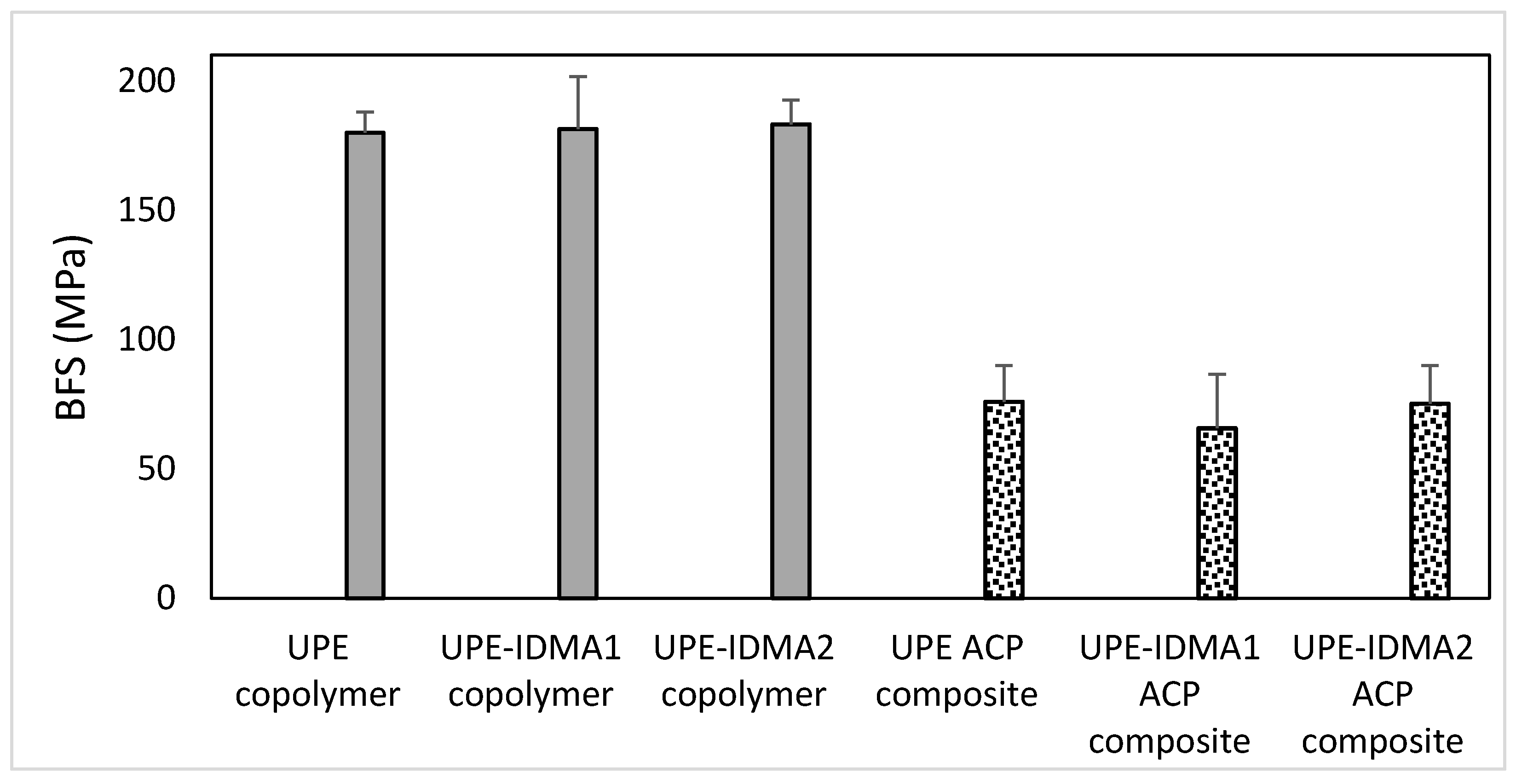
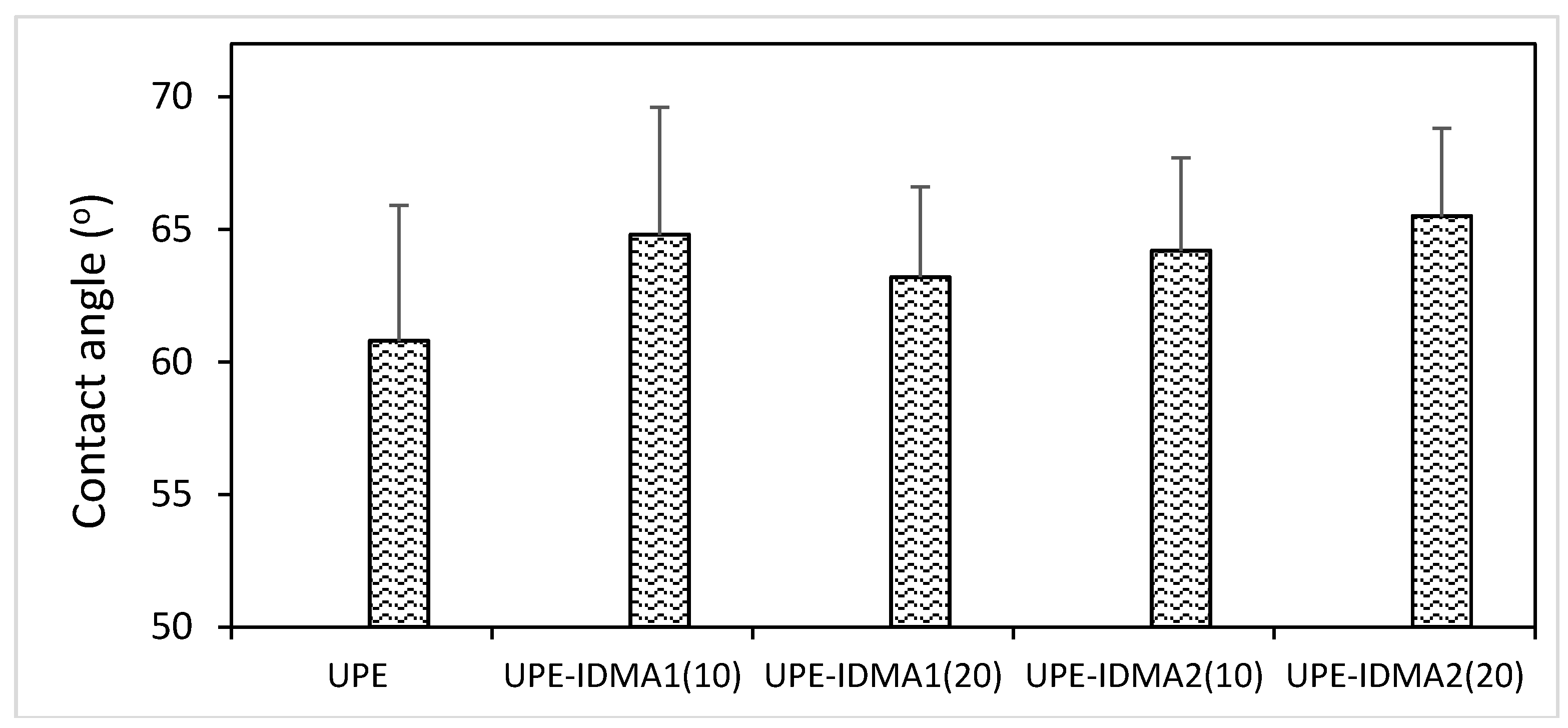
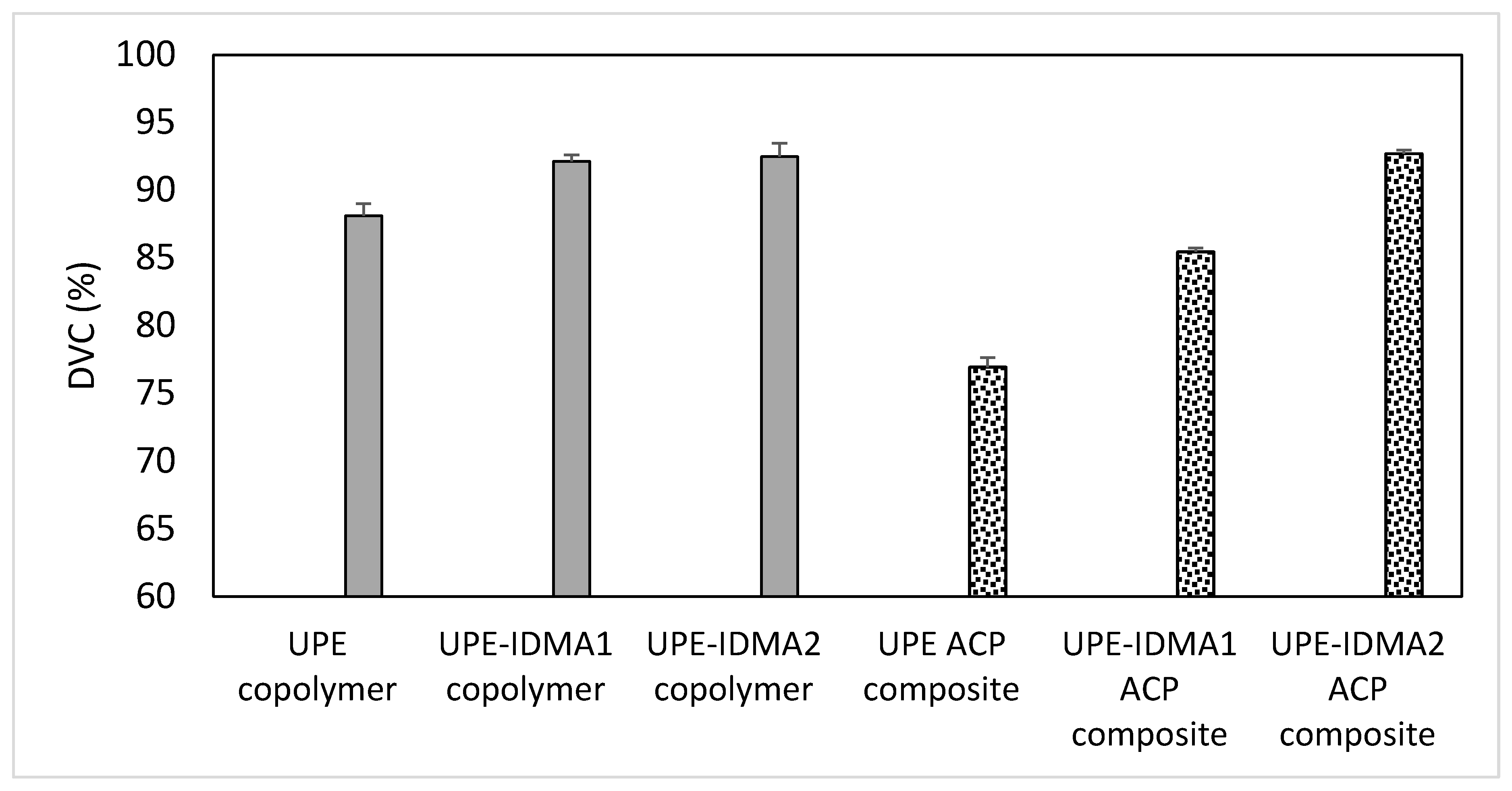
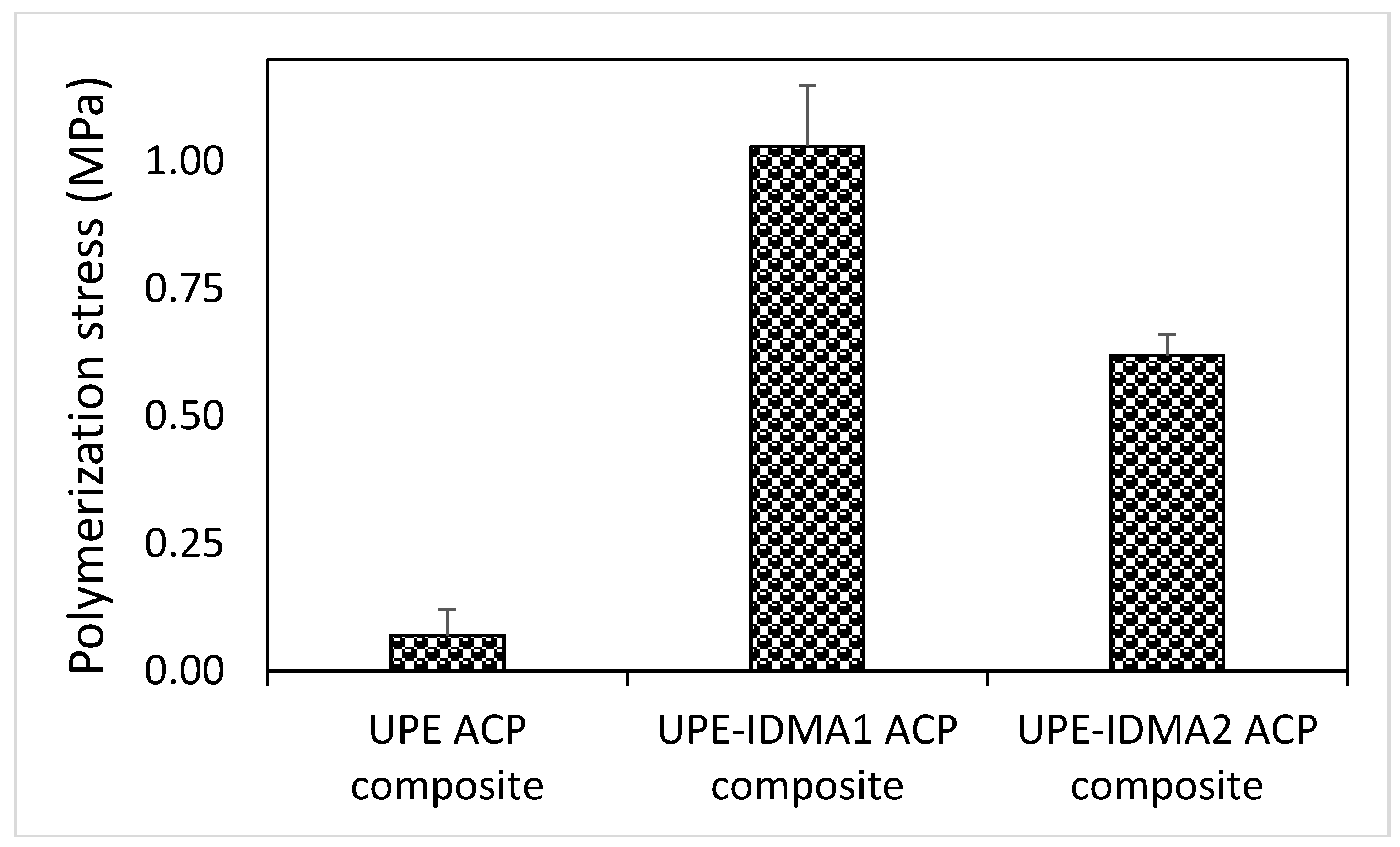
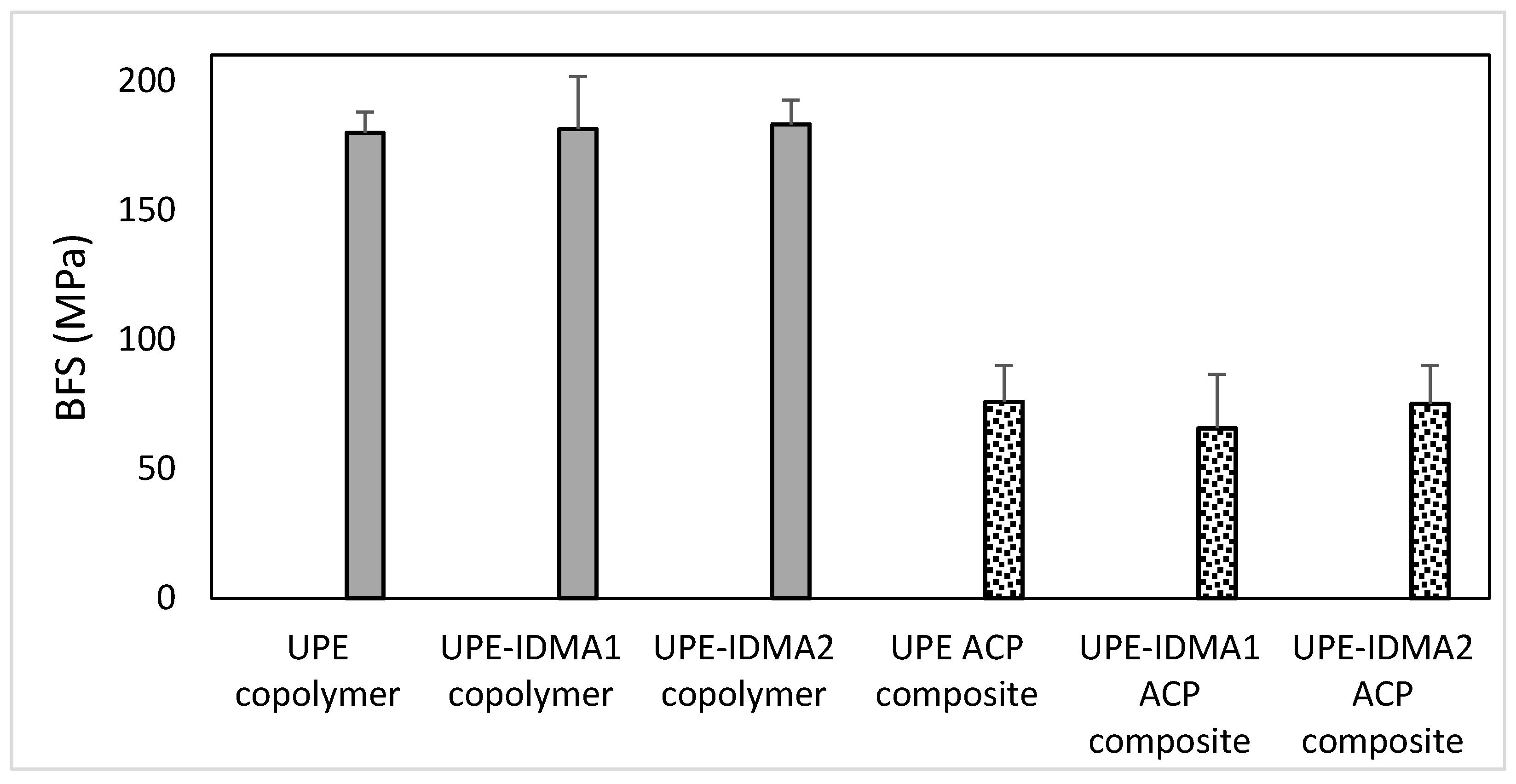
2.4. Preliminary Physicochemical Evaluation of Resins and Composites
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. IDMAs: Syntheses and Purification
4.3. IDMAs: Structural Verification
4.4. Biocompatibility Tests
4.5. Resin Formulation and Evaluation
4.6. Remineralizing ACP Composites
4.6.1. ACP Filler: Synthesis and Characterization
4.6.2. Fabrication of ACP Composites
4.6.3. Ion Release as Predictor of Remineralization Potential of ACP Composites
4.6.4. Simultaneous Assessment of Polymerization Stress and DVC
4.7. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
Disclaimer
Abbreviations
| List of Acronyms | |
| ACP | amorphous calcium phosphate |
| ADA | American Dental Association |
| AM | antimicrobial |
| AMadh | antimicrobial monomer with improved adhesiveness |
| AMcpl | antimicrobial monomer with improved coupling to filler |
| AMmisc | antimicrobial monomer with improved miscibility |
| AMRE | antimicrobial and remineralizing |
| ANOVA | analysis of variance |
| BEMA | 2-bromoethyl methacrylate |
| BFS | biaxial flexural strength |
| Bis-GMA | 2,2-bis[p-(2-hydroxy-3-methacryloxypropoxy)phenyl]propane |
| Ca | calcium |
| CQ | camphorquinone |
| dm | median diameter |
| DVC | degree of vinyl conversion |
| 4EDMAB | ethyl-4-N,N-dimethylamino benzoate |
| EHMA | ethyl 2-(hydroxymethyl)acrylate |
| FTIR | Fourier-transform infrared spectroscopy |
| HEMA | 2-hydroxyethyl methacrylate |
| HGF | human gingival fibroblast |
| HSQC | heteronuclear single quantum coherence |
| IDMA1 | 2-(methacryloyloxy)-N-(2-(methacryloyloxy)ethyl)-N,N-dimethylethan-1-aminium bromide |
| IDMA2 | N,N′-([1,1′-biphenyl]-2,2′-diylbis(methylene))bis(2-(methacryloyloxy)-N,N-dimethylethan-1-aminium) bromide |
| MS | mass spectroscopy |
| n | number of specimens (or repetitive experiments) |
| NIR | near infra-red |
| NMR | nuclear magnetic resonance |
| P | phosphorus |
| PEG-U | poly(ethylene glycol) extended urethane dimethacrylate |
| pH | -log (molar concentration of hydrogen ions) |
| PO4 | phosphate |
| PSD | particle size distribution |
| QADM | quaternary ammonium dimethacrylates |
| SD | standard deviation |
| SEM | standard error of the mean |
| TEGDMA | triethylene glycol dimethacrylate |
| UDMA | urethane dimethacrylate |
| UPE | UDMA/PEG-U/EHMA resin |
| XRD | X-ray diffraction |
References
- Imazato, S. Antibacterial properties of resin composites and dentin bonding systems. Dent. Mater. 2003, 19, 449–457. [Google Scholar] [CrossRef]
- Imazato, S. Bio-active restorative materials with antibacterial effects: New dimension of innovation in restorative dentistry. Dent. Mater. J. 2009, 28, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, N.J.; Cai, F.; Huq, N.L.; Burrow, M.F.; Reynolds, E.C. New approaches to enhanced remineralization of tooth enamel. J. Dent. Res. 2010, 89, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Mickenautsch, S.; Mount, G.; Yengopal, V. Therapeutic effect of glass-ionomers: An overview of evidence. Aust. Dent. J. 2011, 56, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.A.; Lussi, A.; Seemann, R.; Neuhaus, K.W. Prevention of crown and root caries in adults. Periodontology 2000 2011, 55, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M. Dental composites: Bioactive polymeric amorphous calcium phosphate-based. In Encyclopedia of Biomedical Polymers and Polymeric Biomaterials; Taylor and Francis: New York, NY, USA, 2015; pp. 2443–2462. [Google Scholar]
- Antonucci, J.M.; Skrtic, D. Fine-tuning of polymeric resins and their interfaces with amorphous calcium phosphate. A strategy for designing effective remineralizing dental composites. Polymers 2010, 2, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Antonucci, J.M. Bioactive polymeric composites for tooth mineral regeneration: Physicochemical and cellular aspects. J. Funct. Biomat. 2011, 2, 271–307. [Google Scholar] [CrossRef] [PubMed]
- Skrtic, D.; Hailer, A.W.; Takagi, S.; Antonucci, J.M.; Eanes, E.D. Quantitative assessment of the efficacy of amorphous calcium phosphate/methacrylate composites in remineralizing caries-like lesions artificially produced in bovine enamel. J. Dent. Res. 1996, 75, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Langhorst, S.E.; O’Donnell, J.N.; Skrtic, D. In vitro remineralization of enamel by polymeric amorphous calcium phosphate composite: Quantitative microradiographic study. Dent. Mater. 2009, 25, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Beck-Tan, N.C.; Dhurjati, P.; van Dyk, T.K.; LaRossa, R.A.; Cooper, S.L. Quaternary ammonium functionalized poly (propylene imine) dendrimers as effective antimicrobials: Structure–activity studies. Biomacromolecules 2000, 1, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Koepsel, R.R.; Matyjaszewski, K.; Russell, A.J. Permanent, non-leaching antibacterial surface—2: How high density cationic surfaces kill bacterial cells. Biomaterials 2007, 28, 4870–4879. [Google Scholar] [CrossRef] [PubMed]
- Gottenbos, B.; van der Mei, H.C.; Klatter, F.; Nieuwenhuis, P.; Busscher, H.J. In vitro and in vivo antimicrobial activity of covalently coupled quaternary ammonium silane coatings on silicone rubber. Biomaterials 2002, 23, 1417–1423. [Google Scholar] [CrossRef]
- Lee, S.B.; Koepsel, R.R.; Morley, S.W.; Matyjaszewski, K.; Sun, Y.; Russell, A.J. Permanent, nonleaching antibacterial surfaces. 1. Synthesis by atom transfer radical polymerization. Biomacromolecules 2004, 5, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wu, D.; Fu, R. Studies on the synthesis and antibacterial activities of polymeric quaternary ammonium salts from dimethylaminoethyl methacrylate. React. Funct. Polym. 2007, 67, 355–366. [Google Scholar] [CrossRef]
- Ikeda, T.; Hirayama, H.; Yamaguchi, H.; Tazuke, S.; Watanabe, M. Polycationic biocides with pendant active groups: Molecular weight dependence of antibacterial activity. Antimicrob. Agents Chemother. 1986, 30, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Yamaguchi, H.; Tazuke, S. Molecular weight dependence of antibacterial activity in cationic disinfectants. J. Bioact. Compat. Polym. 1990, 5, 31–41. [Google Scholar] [CrossRef]
- Kenawy, E.-R.; Worley, S.; Broughton, R. The chemistry and applications of antimicrobial polymers: A state-of-the-art review. Biomacromolecules 2007, 8, 1359–1384. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Hua, L.; Ogata, T.; Kurihara, S. Synthesis of water-soluble thermosensitive polymers having phosphonium groups from methacryloyloxyethyl trialkyl phosphonium chlorides–N-isopropylacrylamide copolymers and their functions. J. Appl. Polym. Sci. 2003, 87, 386–393. [Google Scholar] [CrossRef]
- Thebault, P.; Taffin de Givenchy, E.; Levy, R.; Vandenberghe, Y.; Guittard, F.; Geribaldi, S. Preparation and antimicrobial behaviour of quaternary ammonium thiol derivatives able to be grafted on metal surfaces. Eur. J. Med. Chem. 2009, 44, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Ebi, N.; Imazato, S.; Noiri, Y.; Ebisu, S. Inhibitory effects of resin composite containing bactericide-immobilized filler on plaque accumulation. Dent. Mater. 2001, 17, 485–491. [Google Scholar] [CrossRef]
- Imazato, S.; Tarumi, H.; Kato, S.; Ebisu, S. Water sorption and colour stability of composites containing the antibacterial monomer MDPB. J. Dent. 1999, 27, 279–283. [Google Scholar] [CrossRef]
- Thome, T.; Mayer, M.P.; Imazato, S.; Geraldo-Martins, V.R.; Marques, M.M. In vitro analysis of inhibitory effects of the antibacterial monomer MDPB-containing restorations on the progression of secondary root caries. J. Dent. 2009, 37, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Weir, M.D.; Chen, J.; Xu, H.H. Comparison of quaternary ammonium-containing with nano-silver-containing adhesive in antibacterial properties and cytotoxicity. Dent. Mater. 2013, 29, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.A.; Guedes, S.F.; Xu, H.H.; Rodrigues, L.K. Nanotechnology-based restorative materials for dental caries management. Trends Biotechnol. 2013, 31, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Zeiger, D.N.; Tang, K.; Lin-Gibson, S.; Fowler, B.O.; Lin, N.J. Synthesis and characterization of dimethacrylates containing quaternary ammonium functionalities for dental applications. Dent. Mater. 2012, 28, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, K.; Weir, M.D.; Melo, M.A.; Zhou, X.; Xu, H.H. Nanotechnology strategies for antibacterial and remineralizing composites and adhesives to tackle dental caries. Nanomedicine 2015, 10, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, K.; Zhou, C.C.; Weir, M.D.; Zhou, X.D.; Xu, H.H. One-year water-ageing of calcium phosphate composite containing nano-silver and quaternary ammonium to inhibit biofilms. Int. J. Oral Sci. 2016, 8, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Weir, M.D.; Xu, H.H.; Antonucci, J.M.; Kraigsley, A.M.; Lin, N.J.; Lin-Gibson, S.; Zhou, X. Antibacterial amorphous calcium phosphate nanocomposites with a quaternary ammonium dimethacrylate and silver nanoparticles. Dent. Mater. 2012, 28, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Winstead, A.; Hart, K.; Hijji, Y.M.; Jasinski, J.P.; Butcher, R.J. 1-(5-Carboxy-pent-yl)-2,3,3-trimethyl-3H-indol-1-ium bromide monohydrate. Acta Crystallogr. E 2009, 66, o171–o172. [Google Scholar] [CrossRef] [PubMed]
- Bienek, D.R.; Frukhtbeyn, S.A.; Giuseppetti, A.A.; Okeke, U.C.; Pires, R.M.; Antonucci, J.M.; Skrtic, D. Physiochemical characterization and biocompatibility of ionic dimethacrylates for dental restorations. Adv. Mater. Sci. Eng. 2018, submitted. [Google Scholar]
- Antonucci, J.M.; Fowler, B.O.; Weir, M.D.; Skrtic, D.; Stansbury, J.W. Effect of ethyl-alpha-hydroxymethylacrylate on selected properties of copolymers and ACP resin composites. J. Mater. Sci. Mater. Med. 2008, 19, 3263–3271. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Skrtic, D. Bioactive and biocompatible polymeric composites based on amorphous calcium phosphate. In Integrated Biomaterials for Medical Applications; Ramalingam, M., Tiwari, A., Ramakrishna, S., Kobayashi, H., Eds.; Scrivener Publishing: Salem, MA, USA, 2012; pp. 67–119. [Google Scholar]
- Skrtic, D.; Antonucci, J.M. Dental composites based on amorphous calcium phosphate—Resin composition/physicochemical properties study. J. Biomater. Appl. 2007, 21, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Watanabe, T.; Kakihana, M. Internal rotation of biphenyl in solution studied by IR and NMR spectra. J. Phys. Chem. 1986, 90, 1752–1755. [Google Scholar] [CrossRef]
- Mazzanti, A.; Lunazzi, L.; Minzoni, M.; Anderson, J.E. Rotation in biphenyls with a single ortho-substituent. J. Org. Chem. 2006, 71, 5474–5481. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Regnault, W.F.; Skrtic, D. Polymerization shrinkage and stress development in amorphous calcium phosphate/urethane dimethacrylate polymeric composites. J. Compos. Mater. 2010, 44, 355. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.M.; Almeida, G.S.; Poskus, L.T.; Guimaraes, J.G. Relationship between the degree of conversion, solubility and salivary sorption of a hybrid and a nanofilled resin composite. J. Appl. Oral Sci. 2008, 16, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Liu, W.; Hao, Z.; Wu, X.; Yin, J.; Panjiyar, A.; Liu, X.; Shen, J.; Wang, H. Characterization of a low shrinkage dental composite containing bismethylene spiroorthocarbonate expanding monomer. Int. J. Mol. Sci. 2014, 15, 2400–2412. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.R.; Ferracane, J.L. Contraction stress related to degree of conversion and reaction kinetics. J. Dent. Res. 2002, 81, 114–118. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.N.; Langhorst, S.E.; Fow, M.D.; Antonucci, J.M.; Skrtic, D. Light-cured dimethacrylate-based resins and their composites: Comparative study of mechanical strength, water sorption and ion release. J. Bioact. Compat. Polym. 2008, 23, 207–226. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Landis, F.A.; Giuseppetti, A.A.; Lin-Gibson, S.; Chiang, M.Y. Simultaneous measurement of polymerization stress and curing kinetics for photo-polymerized composites with high filler contents. Dent. Mater. 2014, 30, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chiang, M.Y. Correlation between polymerization shrinkage stress and C-factor depends upon cavity compliance. Dent. Mater. 2016, 32, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chiang, M.Y. System compliance dictates the effect of composite filler content on polymerization shrinkage stress. Dent. Mater. 2016, 32, 551–560. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Dental Material | Experimental Groups | Fold Decrease 1 |
|---|---|---|---|
| [28] | Composite BisGMA-TEGDMA | Control: Commercial composite | |
| 20% nACP + 6% QADM (aged 1 day) | 1.36 | ||
| 20% nACP + 0.1% nAg (aged 1 day) | 1.75 | ||
| 20% nACP + 6% QADM + 0.1% nAg (aged 1 day) | 2.69 | ||
| [29] | Composite BisGMA-TEGDMA | Control: Composite (no fluoride) | |
| 19.5% nACP | 1.25 | ||
| 19.5% nACP + 7% QADM | 1.73 | ||
| 19.5% nACP + 0.028% nAg | 1.98 | ||
| 19.5% nACP + 7% QADM + 0.028% nAg | 3.00 | ||
| [24] | Primer/adhesive Primer: HEMA-acrylic/itaconic acids Adhesive: BisGMA-HEMA | Primer control + Adhesive control | |
| Primer w/10% QADM + Adhesive w/10% QADM | 1.65 | ||
| Primer w/0.05% nAg + Adhesive w/0.05% nAg | 2.38 | ||
| [25] | Primer/adhesive Primer: PMGDM-HEMA Adhesive: BisGMA-TEGDMA | Primer control + Adhesive control | |
| Primer w/0.1% nAg + Adhesive control | 2.70 | ||
| Primer w/0.1% nAg + Adhesive w/0.1% nAg, 10% QADM | 2.86 | ||
| Primer w/0.1% nAg + Adhesive w/0.1% nAg, 10% QADM, 10% nACP | 3.07 | ||
| Primer w/0.1% nAg + Adhesive w/0.1% nAg, 10% QADM, 20% nACP | 2.98 | ||
| Primer w/0.1% nAg + Adhesive w/0.1% nAg, 10% QADM, 30% nACP | 4.57 | ||
| Primer w/0.1% nAg + Adhesive w/0.1% nAg, 10% QADM, 40% nACP | 5.11 |
| Atom # | 13C Chemical Shift, ppm | 1H Chemical Shift, ppm | # H`s | Signal Splitting |
|---|---|---|---|---|
| 1 | 18.2 | 1.95 | 6 | singlet |
| 2 | 135.0 | 0 | ||
| 3 | 127.4 | 5.67, 6.15 | 2, 2 | singlets |
| 4 | 166.2 | 0 | ||
| 5 | 58.2 | 4.71 | 4 | multiplet |
| 6 | 63.6 | 4.31 | 4 | multiplet |
| 7, 8 | 52.5 | 3.63 | 6 | singlet |
| Atom # | 13C Chemical Shift, ppm | 1H Chemical Shift, ppm | # H`s | Signal Splitting |
|---|---|---|---|---|
| 1 | 17.9 | 1.87 | 6 | singlet |
| 2 | 135.3 | 0 | ||
| 3 | 126.7 | 5.75, 6.03 | 2, 2 | singlets |
| 4 | 165.6 | 0 | ||
| 5 | 64.7 | 4.04, 4.63 | 2, 2 | doublets |
| 6 | 62.2 | 3.30, 3.59 | 2, 2 | multiplets |
| 7, 8 | 49.9, 50.0 | 2.66, 2.93 | 6, 6 | singlets |
| 9 | 57.6 | 4.22 | 4 | broad |
| 10 | 141.8 | 0 | ||
| 11 | 125.1 | 0 | ||
| 12 | 135.1 | 7.82 | 2 | multiplet |
| 13 | 131.0 | 7.69 | 2 | triplet |
| 14 | 128.4 | 7.63 | 2 | triplet |
| 15 | 132.8 | 7.52 | 2 | multiplet |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bienek, D.R.; Frukhtbeyn, S.A.; Giuseppetti, A.A.; Okeke, U.C.; Skrtic, D. Antimicrobial Monomers for Polymeric Dental Restoratives: Cytotoxicity and Physicochemical Properties. J. Funct. Biomater. 2018, 9, 20. https://doi.org/10.3390/jfb9010020
Bienek DR, Frukhtbeyn SA, Giuseppetti AA, Okeke UC, Skrtic D. Antimicrobial Monomers for Polymeric Dental Restoratives: Cytotoxicity and Physicochemical Properties. Journal of Functional Biomaterials. 2018; 9(1):20. https://doi.org/10.3390/jfb9010020
Chicago/Turabian StyleBienek, Diane R., Stanislav A. Frukhtbeyn, Anthony A. Giuseppetti, Ugochukwu C. Okeke, and Drago Skrtic. 2018. "Antimicrobial Monomers for Polymeric Dental Restoratives: Cytotoxicity and Physicochemical Properties" Journal of Functional Biomaterials 9, no. 1: 20. https://doi.org/10.3390/jfb9010020
APA StyleBienek, D. R., Frukhtbeyn, S. A., Giuseppetti, A. A., Okeke, U. C., & Skrtic, D. (2018). Antimicrobial Monomers for Polymeric Dental Restoratives: Cytotoxicity and Physicochemical Properties. Journal of Functional Biomaterials, 9(1), 20. https://doi.org/10.3390/jfb9010020