Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations



 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. HPMC MBGs Formulation
2.2. Scanning Electron Microscopy (SEM) Measurements
2.3. Electron Paramagnetic Resonance (EPR) Measurements
2.3.1. Spin-Probing
2.3.2. Spin-Labeling Lipase
2.3.3. Interpretation of the EPR Data
2.4. Small-Angle X-ray Scattering (SAXS) Measurements
3. Results
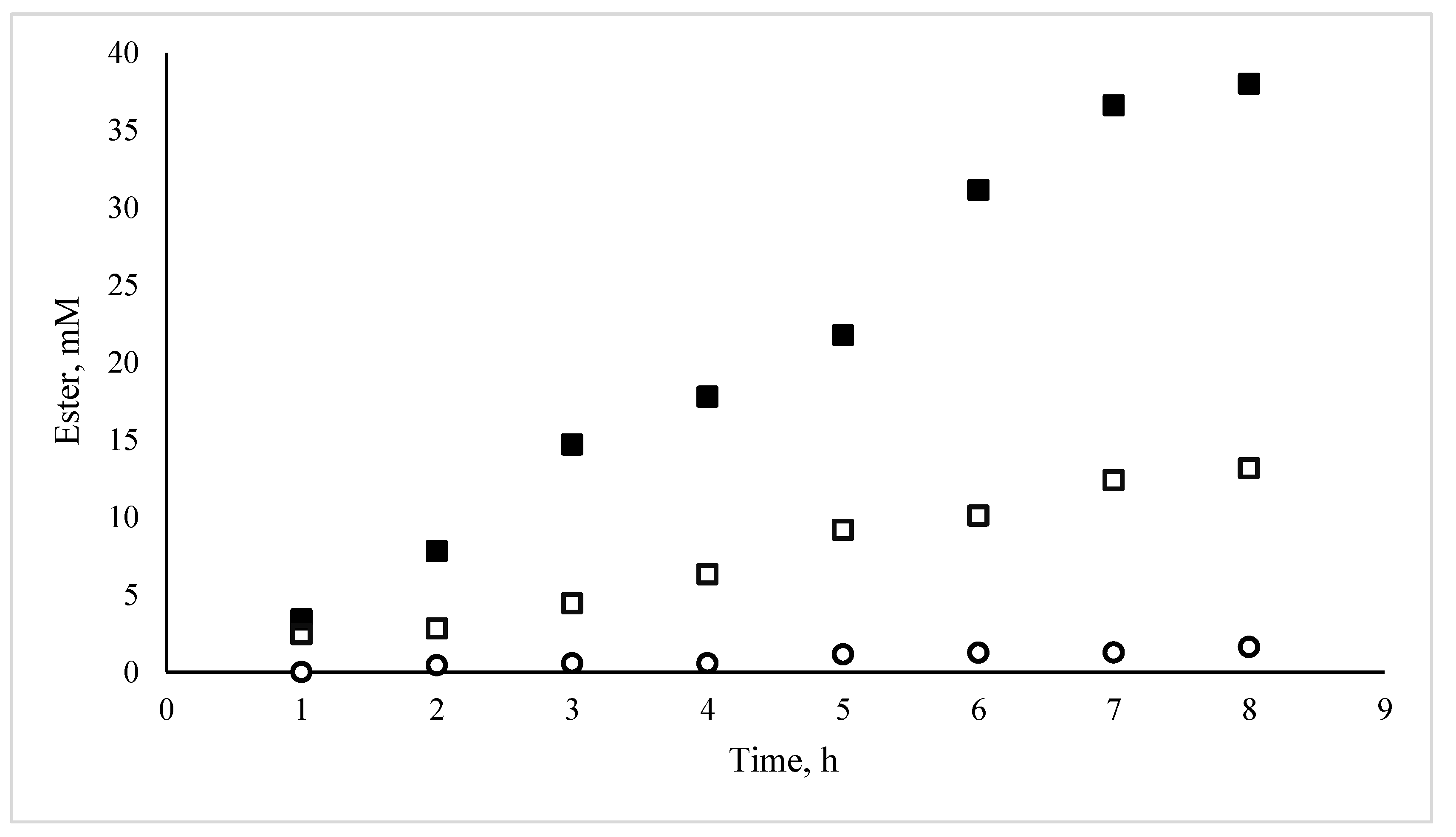
3.1. Catalytic Activity
3.2. Morphological Analysis
3.3. Interfacial Properties
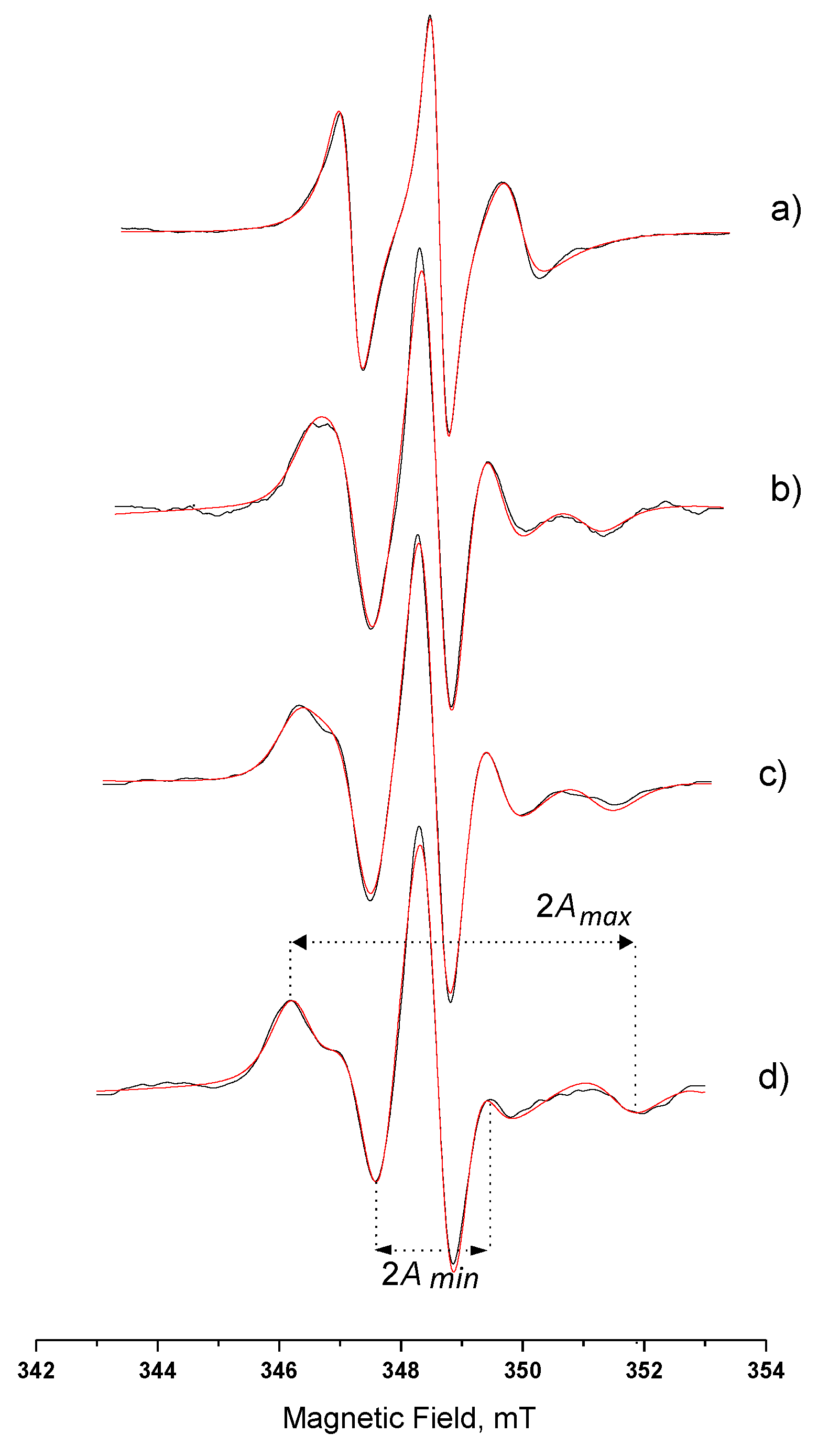
3.3.1. Hydrophilic Spin-Probe
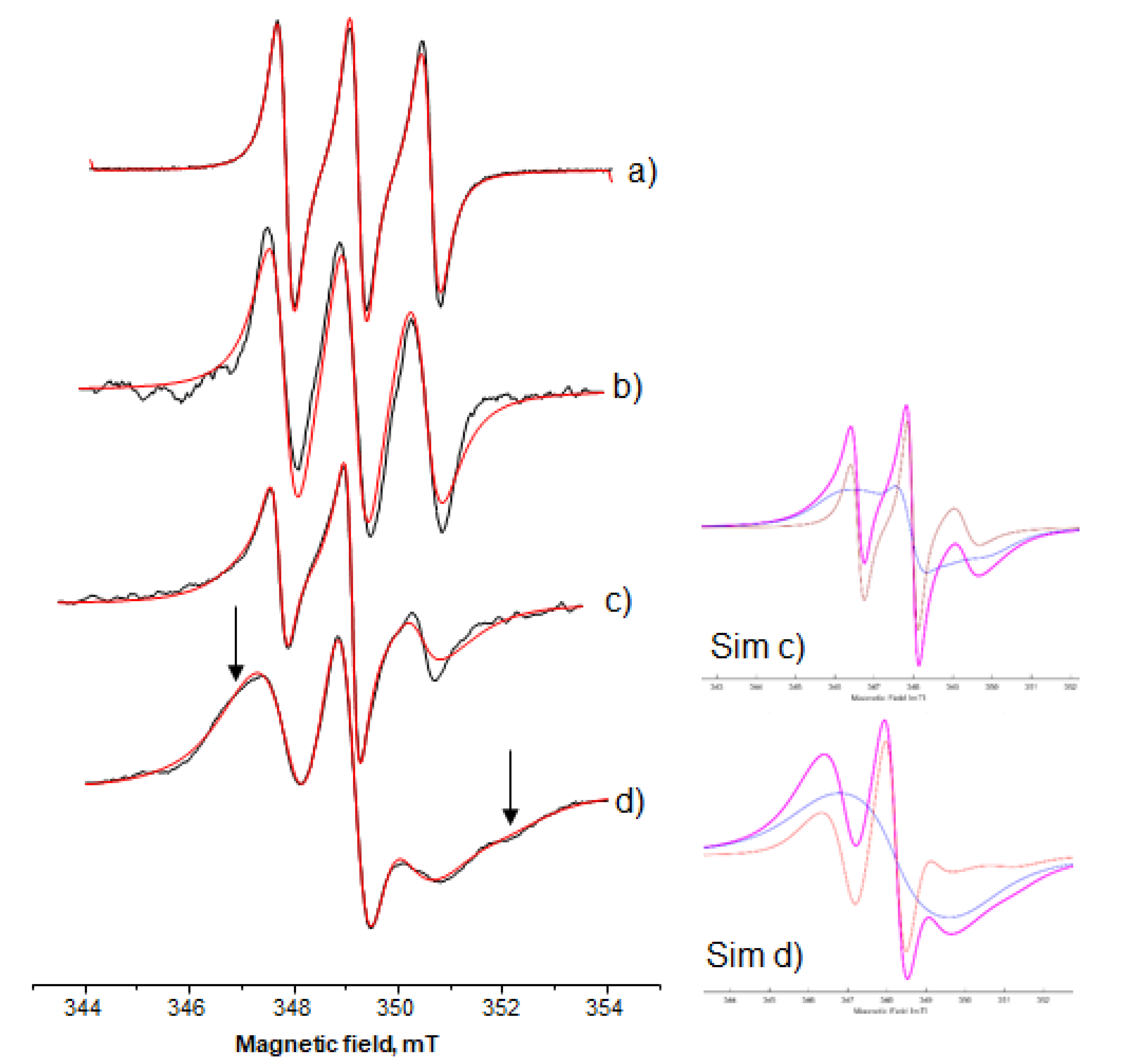
3.3.2. Amphiphilic Spin-Probes
3.3.3. Lipophilic Spin-Probes
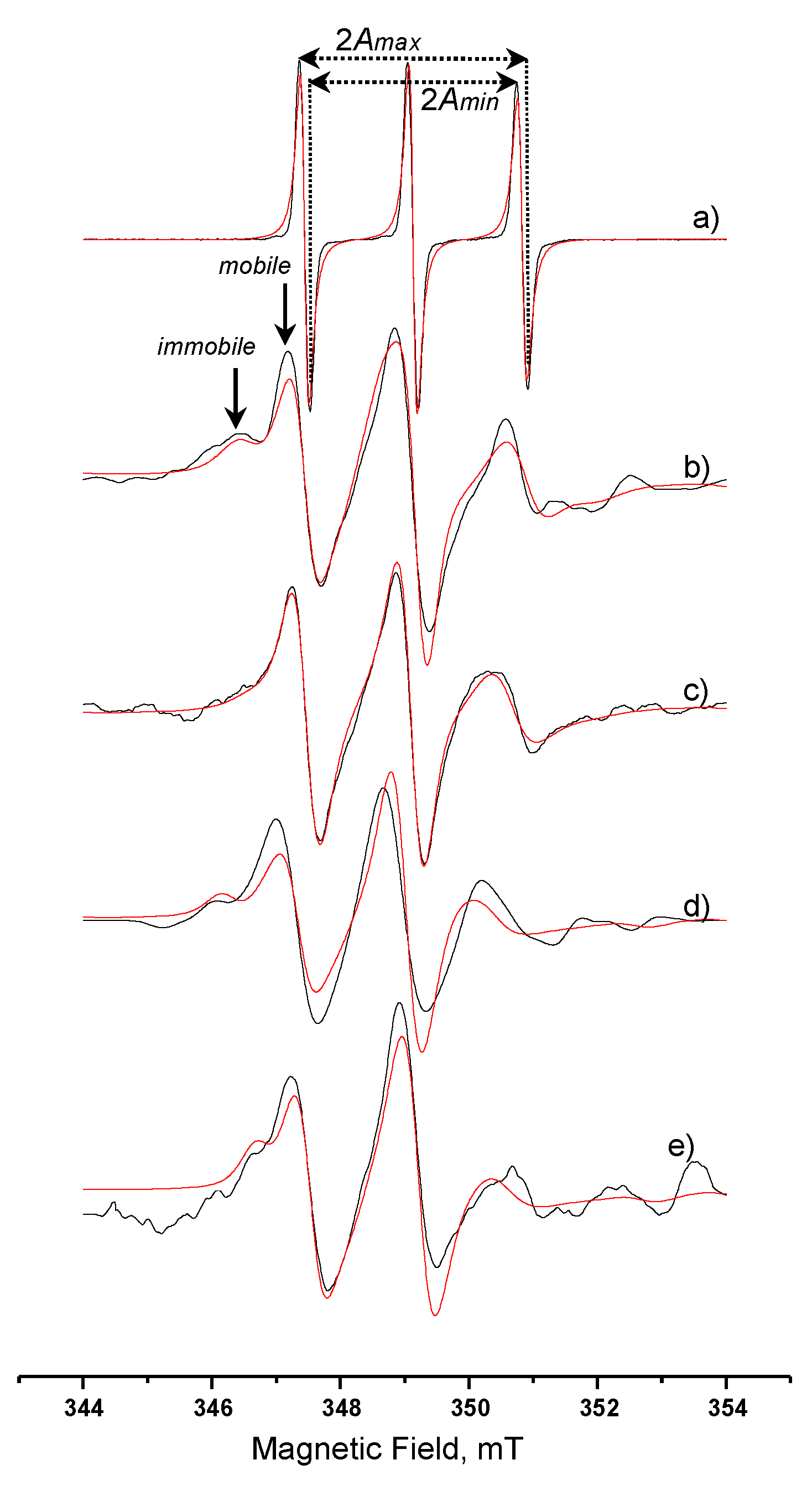
3.3.4. Spin-Labelled Lipase
3.4. Small-Angle X-ray Scattering (SAXS) Measurements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels 2017, 3, 6. [Google Scholar] [CrossRef]
- Zoumpanioti, M.; Karavas, E.; Skopelitis, C.; Stamatis, H.; Xenakis, A. Lecithin organogels as model carriers of pharmaceuticals. Progr. Colloid Polym. Sci. 2004, 123, 199–202. [Google Scholar]
- Avramiotis, S.; Papadimitriou, V.; Hatzara, E.; Bekiari, V.; Lianos, P.; Xenakis, A. Lecithin organogels used as bioactive compounds carriers. A microdomain properties investigation. Langmuir 2007, 23, 4438–4447. [Google Scholar] [CrossRef]
- De Souza Paglarini, C.; de Figueiredo Furtado, G.; Biachi, J.P.; Vidal, V.A.S.; Martini, S.; Forte, M.B.S.; Cunha, R.L.; Pollonio, M.A.R. Functional emulsion gels with potential application in meat products. J. Food Eng. 2018, 222, 29–37. [Google Scholar] [CrossRef]
- Huang, Y.; Mei, L.; Chen, X.; Wang, Q. Recent developments in food packaging based on nanomaterials. Nanomaterials 2018, 8, 830. [Google Scholar] [CrossRef]
- Kim, M.H.; An, S.; Won, K.; Kim, H.J.; Lee, S.H. Entrapment of enzymes into cellulose-biopolymer composite hydrogel beads using biocompatible ionic liquid. J. Mol. Catal. B Enzym. 2012, 75, 68–72. [Google Scholar] [CrossRef]
- Vassiliadi, E.; Xenakis, A.; Zoumpanioti, M. Chitosan hydrogels: A new and simple matrix for lipase catalysed biosyntheses. Mol. Catal. 2018, 445, 206–212. [Google Scholar] [CrossRef]
- Laurienzo, P.; Malinconico, M.; Pizzano, R.; Manzo, C.; Piciocchi, N.; Sorrentino, A.; Volpe, M.G. Natural polysaccharide-based gels for dairy food preservation. J. Dairy Sci. 2006, 89, 2856–2864. [Google Scholar] [CrossRef]
- Bilal, M.; Iqbal, H.M.N. Naturally-derived biopolymers: Potential platforms for enzyme immobilization. Int. J. Biol. Macromol. 2019, 130, 462–482. [Google Scholar] [CrossRef]
- Turner, M.B.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Production of bioactive cellulose films reconstituted from ionic liquids. Biomacromolecules 2004, 5, 1379–1384. [Google Scholar] [CrossRef]
- Dalla-Vecchia, R.; Sebrão, D.; Nascimento, M.D.G.; Soldi, V. Carboxymethylcellulose and poly(vinyl alcohol) used as a film support for lipases immobilization. Process Biochem. 2005, 40, 2677–2682. [Google Scholar] [CrossRef]
- Arboleya, J.C.; Wilde, P.J. Competitive adsorption of proteins with methylcellulose and hydroxypropyl methylcellulose. Food Hydrocoll. 2005, 19, 485–491. [Google Scholar] [CrossRef]
- Pérez, O.E.; Sánchez, C.C.; Pilosof, A.M.R.; Rodríguez Patino, J.M. Kinetics of adsorption of whey proteins and hydroxypropyl-methyl-cellulose mixtures at the air-water interface. J. Colloid Interface Sci. 2009, 336, 485–496. [Google Scholar] [CrossRef]
- Rodriguez Patino, J.M.; Pilosof, A.M.R. Protein-polysaccharide interactions at fluid interfaces. Food Hydrocoll. 2011, 25, 1925–1937. [Google Scholar] [CrossRef]
- Pérez, O.E.; Carrera Sánchez, C.; Pilosof, A.M.R.; Rodríguez Patino, J.M. Impact of hydroxypropylmethylcellulose on whey protein concentrate spread film at the air-water interface: Structural and surface dilatational characteristics. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 465, 1–10. [Google Scholar] [CrossRef]
- Fathi, M.; Martín, Á.; McClements, D.J. Nanoencapsulation of food ingredients using carbohydrate based delivery systems. Trends Food Sci. Technol. 2014, 39, 18–39. [Google Scholar] [CrossRef]
- Raghavendra, T.; Sayania, D.; Madamwar, D. Synthesis of the “green apple ester” ethyl valerate in organic solvents by Candida rugosa lipase immobilized in MBGs in organic solvents: Effects of immobilization and reaction parameters. J. Mol. Catal. B Enzym. 2010, 63, 31–38. [Google Scholar] [CrossRef]
- Nagayama, K.; Karaiwa, K.; Doi, T.; Imai, M. Esterification activity and stability of Candida rugosa lipase in AOT microemulsion-based organogels. Biochem. Eng. J. 1998, 2, 121–126. [Google Scholar] [CrossRef]
- Quellet, C.; Eicke, H.-F. Mutual gelation of gelatin and water-in-oil microemulsions. Chimia 1986, 40, 233–238. [Google Scholar]
- Haering, G.; Luisi, P.L. Hydrocarbon Gels from Water-in-Oil Microemulsions. J. Phys. Chem. 1986, 16, 5892–5895. [Google Scholar] [CrossRef]
- Pastou, A.; Stamatis, H.; Xenakis, A. Microemulsion-based organogels containing lipase: Application in the synthesis of esters. Progr. Colloid Polym. Sci. 2000, 115, 192–195. [Google Scholar]
- Delimitsou, C.; Zoumpanioti, M.; Xenakis, A.; Stamatis, H. Activity and stability studies of Mucor miehei lipase immobilized in novel microemulsion-based organogels. Biocatal. Biotransform. 2002, 20, 319–327. [Google Scholar] [CrossRef]
- Zoumpanioti, M.; Merianou, E.; Karandreas, T.; Stamatis, H.; Xenakis, A. Esterification of phenolic acids catalyzed by lipases immobilized in organogels. Biotechnol. Lett. 2010, 32, 1457–1462. [Google Scholar] [CrossRef]
- Zoumpanioti, M.; Stamatis, H.; Xenakis, A. Microemulsion-based organogels as matrices for lipase immobilization. Biotechnol. Adv. 2010, 28, 395–406. [Google Scholar] [CrossRef]
- Griffith, O.H.; Jost, P.C. Lipid Spin Labels in Biological Membranes. In Spin Labeling; Academic Press: Cambridge, MA, USA, 1976; Volume 1, pp. 453–523. [Google Scholar]
- Papadimitriou, V.; Sotiroudis, T.G.; Xenakis, A. Olive Oil Microemulsions: Enzymatic Activities and Structural Characteristics. Langmuir 2007, 23, 2071–2077. [Google Scholar] [CrossRef]
- Fanun, M.; Papadimitriou, V.; Xenakis, A. Characterization of Cephalexin Loaded Nonionic Microemulsions. J. Colloid Interface Sci. 2011, 361, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Knauer, B.R.; Napier, J.J. The Nitrogen Hyperfine Splitting Constant of the Nitroxide Functional Group as a Solvent Polarity Parameter. The Relative Importance for a Solvent Polarity Parameter of Its Being a Cybotactic Probe vs. Its Being a Model Process. J. Am. Chem. Soc. 1976, 98, 4395–4400. [Google Scholar] [CrossRef]
- Marsh, D. Spin-Label EPR for Determining Polarity and Proticity in Biomolecular Assemblies: Transmembrane Profiles. Appl. Magn. Reson. 2010, 37, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Hemminga, M.A.; Berliner, L. ESR Spectroscopy in Membrane Biophysics; Biological Magnetic Resonance; Springer US: Boston, MA, USA, 2007; Volume 27. [Google Scholar]
- Etienne, E.; Le Breton, N.; Martinho, M.; Mileo, E.; Belle, V. SimLabel: A graphical user interface to simulate continuous wave EPR spectra from site-directed spin labeling experiments. Magn. Reson. Chem. 2017, 55, 714–719. [Google Scholar] [CrossRef]
- Zoumpanioti, M.; Karali, M.; Xenakis, A.; Stamatis, H. Lipase biocatalytic processes in surfactant free microemulsion-like ternary systems and related organogels. Enzym. Microb. Technol. 2006, 39, 531–539. [Google Scholar] [CrossRef]
- Stamatis, H.; Xenakis, A. Biocatalysis using microemulsion-based polymer gels containing lipase. J. Mol. Catal. B Enzym. 1999, 6, 399–406. [Google Scholar] [CrossRef]
- Itabaiana, I.; Gonçalves, K.M.; Zoumpanioti, M.; Leal, I.C.R.; Miranda, L.S.M.E.; Xenakis, A.; De Souza, R.O.M.A. Microemulsion-based organogels as an efficient support for lipase-catalyzed reactions under continuous-flow conditions. Org. Process Res. Dev. 2014, 18, 1372–1376. [Google Scholar] [CrossRef]
- Blattner, C.; Zoumpanioti, M.; Kröner, J.; Schmeer, G.; Xenakis, A.; Kunz, W. Biocatalysis using lipase encapsulated in microemulsion-based organogels in supercritical carbon dioxide. J. Supercrit. Fluids 2006, 36, 182–193. [Google Scholar] [CrossRef]
- Dandavate, V.; Madamwar, D. Reusability of surfactant-coated Candida rugosa lipase immobilized in gelatin microemulsion-based organogels for ethyl isovalerate synthesis. J. Microbiol. Biotechnol. 2008, 18, 735–741. [Google Scholar]
- Zhang, W.W.; Wang, N.; Zhang, L.; Wu, W.X.; Hu, C.L.; Yu, X.Q. Effects of additives on lipase immobilization in microemulsion-based organogels. Appl. Biochem. Biotechnol. 2014, 172, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Moulik, S.P.; Paul, B.K. Structure, dynamics and transport properties of micro emulsions. Adv. Colloid Interface Sci. 1998, 78, 99–195. [Google Scholar] [CrossRef]
- González-Blanco, C.; Rodríguez, L.J.; Velázquez, M.M. Effect of the solvent on the water properties of water/oil microemulsions. J. Colloid Interface Sci. 1999, 211, 380–866. [Google Scholar] [CrossRef]
- Haering, G.; Luisi, P.L.; Hauser, H. Characterization by electron spin resonance of reversed micelles consisting of the ternary system AOT-isooctane-water. J. Phys. Chem. 1988, 92, 3574–3581. [Google Scholar] [CrossRef]
- Schreier, S.; Polnaszek, C.F.; Smith, I.C.P. Spin labels in membranes problems in practice. BBA Rev. Biomembr. 1978, 515, 395–436. [Google Scholar] [CrossRef]
- Caldararu, H.; Timmins, G.S.; Gilbert, B.C. The Structure of Gelatin-Water/Oil Microemulsion Sols and Gels. An EPR Spin-Probe and Spin-Labelling Study. Phys. Chem. Chem. Phys. 1999, 1, 5689–5695. [Google Scholar] [CrossRef]
- Alberghina, L.; Lotti, M. [14] Cloning, sequencing, and expression of Candida rugosa lipases. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 284, pp. 246–260. [Google Scholar]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Bouthillier, F.; Smith, P.; Harrison, D.; Rubin, B.; Cygler, M. Insights into interfacial activation from an open structure of Candida rugosa lipase. J. Biol. Chem. 1993, 268, 12843–12847. [Google Scholar]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, L.; Vincent, C.; Lei, X.; Ottaviani, M.F.; Ananthapadmanabhan, K.P. A comparison between interactions of triglyceride oil and mineral oil with proteins and their ability to reduce cleanser surfactant-induced irritation. Int. J. Cosmet. Sci. 2015, 37, 371–378. [Google Scholar] [CrossRef]
- Bösecke, P.; Diat, O. Small-angle X-ray scattering at the ESRF high-brilliance beamline. J. Appl. Crystallogr. 1997, 30, 867–871. [Google Scholar] [CrossRef]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Crystallogr. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Hammouda, B.; Ho, D.L.; Kline, S. Insight into clustering in poly(ethylene oxide) solutions. Macromolecules 2004, 37, 6932–6937. [Google Scholar] [CrossRef]
- Bhattacharjee, S.M.; Giacometti, A.; Maritan, A. Flory theory for polymers. J. Phys. Condens. Matter 2013, 25, 503101. [Google Scholar] [CrossRef]
- Atkinson, P.J.; Heenan, R.K.; Grimson, M.J.; Howe, A.M.; Robinson, B.H. Structure of microemulsion-based organo-gels. Progr. Colloid Polym. Sci. 1990, 23, 1807–1809. [Google Scholar]
- Petit, C.; Zemb, T.; Pileni, M.P. Structural Study of Microemulsion-Based Gels at the Saturation Point. Langmuir 1991, 7, 223–231. [Google Scholar] [CrossRef]
- Hauser, H.; Haering, G.; Pande, A.; Luisi, P.L. Interaction of water with sodium bis(2-ethyl-1-hexyl) sulfosuccinate in reversed micelles. J. Phys. Chem. 1989, 93, 7869–7876. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MBG | HPMC | H2O | µE |
|---|---|---|---|
| % w/w | |||
| System A | 18 | 71 | 11 |
| System B | 28 | 55 | 17 |
| System C | 44 | 43 | 13 |
| Hydrophilic Probe Hydroxy-TEMPO | ||
|---|---|---|
| System | τR, ns | AN, 10−4 T |
| Water | 0.03 ± 0.00 | 17.49 ± 0.04 |
| System A* | 0.07 ± 0.01 | 17.29 ± 0.11 |
| System B* | 0.15 ± 0.01 | 17.25 ± 0.03 |
| System C* | 0.40 ± 0.01 | 17.07 ± 0.04 |
| System C** | 0.40 ± 0.06 | 17.07 ± 0.03 |
| (AOT) = 0.1 Μ µE wo = 15 | 0.13 ± 0.01 | 16.25 ± 0.02 |
| (AOT) = 0.2 Μ, µE wo = 7.5 | 0.21 ± 0.01 | 15.84 ± 0.01 |
| Probe incorporated in the HPMC-based MBGs via the AOT microemulsion | ||
| System A | 0.06 ± 0.01 | 17.31 ± 0.01 |
| System B | 0.14 ± 0.01 | 17.21 ± 0.02 |
| System C | 0.40 ± 0.01 | 17.14 ± 0.01 |
| System C† | 0.40 ± 0.06 | 17.11 ± 0.05 |
| Probe incorporated in the HPMC-based MBG via the HPMC/water mixture | ||
| System A | 0.08 ± 0.01 | 17.35 ± 0.04 |
| System B | 0.15 ± 0.02 | 17.22 ± 0.03 |
| System C | 0.38 ± 0.03 | 17.11 ± 0.02 |
| Amphiphilic Probes | ||||||
|---|---|---|---|---|---|---|
| System | 5 DSA | 16 DSA | ||||
| τR, ns | S | AN, 10−4 T | τR, ns | S | AN, 10−4 T | |
| µE wo = 15 | 3.45 ± 0.10 | 0.29 ± 0.02 | 15.06 ± 0.03 | 0.07 ± 0.01 | 0.06 ± 0.01 | 14.39 ± 0.08 |
| μE wo = 7.5 | 2.30 ± 0.08 | 0.14 ± 0.01 | 14.01 ± 0.05 | 0.09 ± 0.02 | 0.09 ± 0.01 | 14.26 ± 0.06 |
| System A | 6.02 ± 0.47 | 0.49 ± 0.03 | 15.08 ± 0.12 | 0.95 ± 0.01 | 0.05 ± 0.01 | 14.97 ± 0.02 |
| System B | 6.57 ± 0.05 | 0.55 ± 0.01 | 15.37 ± 0.05 | 1.39 ± 0.05 | 0.07 ± 0.01 | 14.85 ± 0.01 |
| System C | 6.96 ± 0.14 | 0.61 ± 0.03 | 15.69 ± 0.17 | 3.31 ± 0.04 | 0.17 ± 0.01 | 14.21 ± 0.14 |
| Spin-Labelled Lipase from Candida rugosa—Iodoacetamide Tempo | |||||
|---|---|---|---|---|---|
| τR, ns | S | AN, 10−4T | |||
| Free spin label in water | 0.06 ± 0.01 | 0.01 ± 0.01 | 17.48 ± 0.09 | ||
| Spin-labelled Candida rugosa in aqueous solution | |||||
| Two components | Immobile | – | mobile | ||
| 10.83 ns (44%) | – | 1.66 ns (56%) | 0.41 ± 0.03 | 17.23 ± 0.14 | |
| Spin-labelled Candida rugosa in AOT microemulsion, wo = 15 | |||||
| Two components | Immobile | – | mobile | ||
| 8.92 ns (20%) | – | 1.78 ns (80%) | 0.14 ± 0.06 | 16.00 ± 0.03 | |
| Spin-labelled Candida rugosa in HPMC-based MBGs | |||||
| Two components | Immobile | – | mobile | ||
| System A | 19.85 ns (45%) | – | 3.16 ns (55%) | 0.35 ± 0.04 | 16.48 ± 0.13 |
| System B | 21.87 ns (43%) | – | 3.31 ns (57%) | 0.39 ± 0.05 | 16.73 ± 0.08 |
| Parameters | System C* | System C** | System C‡ | System C† |
|---|---|---|---|---|
| Correlation length (Å) | 44.6 | 44.1 | 66.9 | 71.6 |
| Porod exponent (n) | 4.5 | 4.5 | 4.8 | 5.1 |
| Lorentzian exponent (m) | 3.0 | 3.2 | 2.8 | 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassiliadi, E.; Mitsou, E.; Avramiotis, S.; Chochos, C.L.; Pirolt, F.; Medebach, M.; Glatter, O.; Xenakis, A.; Zoumpanioti, M. Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations. Nanomaterials 2020, 10, 2204. https://doi.org/10.3390/nano10112204
Vassiliadi E, Mitsou E, Avramiotis S, Chochos CL, Pirolt F, Medebach M, Glatter O, Xenakis A, Zoumpanioti M. Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations. Nanomaterials. 2020; 10(11):2204. https://doi.org/10.3390/nano10112204
Chicago/Turabian StyleVassiliadi, Evdokia, Evgenia Mitsou, Spyridon Avramiotis, Christos L. Chochos, Franz Pirolt, Martin Medebach, Otto Glatter, Aristotelis Xenakis, and Maria Zoumpanioti. 2020. "Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations" Nanomaterials 10, no. 11: 2204. https://doi.org/10.3390/nano10112204
APA StyleVassiliadi, E., Mitsou, E., Avramiotis, S., Chochos, C. L., Pirolt, F., Medebach, M., Glatter, O., Xenakis, A., & Zoumpanioti, M. (2020). Structural Study of (Hydroxypropyl)Methyl Cellulose Microemulsion-Based Gels Used for Biocompatible Encapsulations. Nanomaterials, 10(11), 2204. https://doi.org/10.3390/nano10112204