1. Introduction
Industry development imply heavy economical charges associated to waste removal, often a cocktail of pollutants harmful to the environment, dangerous for human health, and difficult to degrade by natural mean [
1]. Among common persistent pollutants, phenol derivatives are a ubiquitous group. Common sources of phenols pollution in water bodies are paints, pesticides, coal, polymers, food, cosmetic and pharmaceutical industries, resins, oil and petrochemical products. Phenol is cytotoxic, damaging the skin and mucous membranes when concentrated, while causing erythema, vesicles and ulcerations, when dilute. It may also cause peripheral neuritis, renal malfunction and liver/kidney necrosis. Moreover, phenol acts as a cardiovascular depressant. General intoxication by phenol may be severe, including possible vascular collapse, respiratory failure and death. Thus, EU directive 80/778/CE has limited phenol concentration in drinking water to 0.5 μg/L [
2]. Phenol has been widely accepted as a model persistent organic pollutant in different pollution abatement studies, including photodegradation technologies.
Different methods have been used to achieve phenol safety level, in the range 0.1–1.0 mg·L
−1 [
3]: adsorption, electrochemical oxidation, biological treatment, etc. However, these processes generate byproducts that may be harmful, making additional treatments necessary, implying further costs [
4]. Additionally, these methods cannot give satisfactory yields in terms of pollutant removal because of the solubility of phenol in water. Therefore, it is essential to develop modern technologies for efficient and cheaper treatments [
5]. Photocatalysis is one of the most promising methods for complete mineralization of persistent organic pollutants like phenol and derivatives, thus avoiding generation of secondary pollutants [
1,
4]. In brief, photocatalytic degradation of pollutants involves formation of e
−/h
+ pairs upon irradiation of the semiconductor (SC) with photons of energy higher than or equal to the band gap energy (hυ ≥ Eg). An electron is excited from the valence band (VB) to the conduction band (CB) of the SC, yielding an oxidation site (hole, h
+) and a reduction site (electron, e
−). Holes, h
+, may oxidize adsorbed organic species, water or HO
–, forming strongly oxidant hydroxyl radicals, HO
•, or organic radical cations, R
•+. Electrons, e
−, may reduce O
2 to the superoxide radical anion, O
2–, or organic species to the corresponding radical anions, R
–. Finally, degradation of organic compounds C
xH
yO
2 into CO
2 and H
2O takes place after reaction with HO
•, O
2•–, or breakage of R
+ or R
– [
6].
A number of SCs have been tested in heterogeneous photocatalysis: TiO
2, ZnO, ZnS, WO
3, GaP, Fe
2O
3, CdS, etc. [
7,
8]. The most extensively used photocatalyst is TiO
2 which presents many advantages compared to others: it is abundant, inexpensive, stable, efficient and non-toxic [
9]. The most effective form of TiO
2 for heterogeneous photocatalysis is the commercial titania mixture Evonik TiO
2-P25. Its very high photocatalytic activity is due to the anatase-rutile junction that reduces the rate of e
−/h
+ recombination [
10]. Despite its excellent photocatalytic performance, it shows a number of drawbacks: (i) it requires excitation wavelengths shorter than 415 nm, as the overlap between sunlight emission and TiO
2-P25 absorbance is very low, ca. 4% UV and (ii) e
−/h
+ recombination is large, limiting its photoactivity. Other SCs (e.g., CdS or GaP) have advantages such as absorbing larger fraction of sunlight as compared to TiO
2, however, they undergo photocorrosion during the photocatalytsis. Different strategies have been developed to improve Vis light absorption and/or to reduce e
−/h
+ recombination. Among these, doping and impregnation with transition metal ions lead to an improvement in photocatalytic activity [
11,
12] through the generation of intermediate energy states in the band gap of TiO
2 (increasing Vis light absorption) or trapping of photoexcited electrons (reducing e
−/h
+ recombination) [
13]. Photocatalysts have been used for pollution abatement in water, both in suspension and immobilized over suitable supports. Alternative strategies, such as doping TiO
2 onto large particles avoid the expensive cost of nanofiltration in real-world environmental applications [
14]. Here, we focus on the behavior of suspended photocatalysts, leaving its immobilization for a later stage.
In this study, we have impregnated TiO2-P25 with different amounts of metals (Cu, Cr, and V), to improve visible light harvesting, and investigated the variables controlling phenol photodegradation, as a model of phenolic pollutants abatement, by heterogeneous photocatalysis with the resulting materials under Vis and UV light. 0.1%, 0.3%, 0.5%, and 1% of Cu, Cr, and V were used, and the corresponding reaction mechanism for the phenol photocatalyzed degradation was described.
2. Experimental
2.1. Materials
TiO
2-P25 was purchased from Evonik (
ca. 70:30% anatase: rutile with a small amount of amorphous phase and a surface area of 55 ± 15 m
2·g
−1) [
15]. Copper (II) sulfate pentahydrate (CuSO
4 5H
2O) (≥98%, Sigma), chromium (III) nitrate (Cr(NO
3)
3·9H
2O (≥98%, Sigma), ammonium metavanadate NH
4VO
3 (99.996%, Sigma), and phenol (C
6H
5OH) (99.5% Sigma-Aldrich) were purchased and used without further purification. Acetonitrile was purchased for J.T. Baker with HPLC grade. O
2 (purity ≥ 99.995%) gas was used in some experiments. Distilled water used in the experiments was obtained from a Millipore apparatus (Milli-Q water) with a resistivity of 18.2 MΩ at 298.0 K and total organic carbon (TOC) less than 5 μg·L
−1.
2.2. Catalyst Synthesis
An incipient wetness impregnation method was adopted for metal immobilization. The desired amount of metal salt was dissolved in distilled water, to which 1 g of TiO2-P25 was added. The mixture was then kept under vigorous stirring at 50 °C for 24 h. This suspension was dried at 50 °C. Finally, the photocatalysts were calcined at 500 °C for 4 h with a ramp rate of 100 °C/h. The resulting Cu/TiO2, Cr/TiO2, and V/TiO2 photocatalysts were thoroughly ground and labeled as M(X%)/TiO2 where M stands for the metal and X represents its mass percentage (0.1%, 0.3%, 0.5%, and 1%). To test the stability of the photocatalysts, they were suspended in distilled water for 2 h, with mechanical stirring, and the filtrate composition was analysed for the presence of the corresponding metal cation.
2.3. Characterization Techniques
The surface morphology of TiO2-P25 and the different M(X%)/TiO2 was recorded using Transmission Electron Microscopy (TEM). The samples were prepared by depositing drops of nanoparticle solutions on carbon formvar coated copper grids (electron microscopy, 200 mesh) and air drying. TEM images were obtained with a Jeol JEM 1100 instrument operating at an acceleration voltage of 80 kV the carbon content was studied by elemental analysis (Thermo Flash 1112). X-ray diffraction (XRD) measurements were carried out on a Bruker Siemens D5000 diffractometer with Bragg–Brentano geometry and θ/2θ configuration, equipped with a graphite monochromator. The optics consist of 2° primary and secondary Soller slits, variable output slit, 1 mm reception slit, 0.2 mm monochromator slit and 0.6 mm detector slit. The detector was a scintillation counter. The conditions of acquisition were: sweeping range (2θ): 2–80°, skip size (step size): 0.050°, acquisition time in each jump (time per step) 2.5 s. DiffracPlus v. 8.0.0.2 (Socabim) software was used for data processing.
The anatase mass fraction in the synthesized metal impregnated photocatalysts was calculated from XRD data using the Spurr and Myers Equation (1): [
16]
where
fA is the mass fraction of crystalline anatase in the TiO
2-P25 nanoparticles,
IR and
IA are the intensity of the (110) rutile and of the (101) anatase reflection, respectively.
The Scherrer Equation (2): [
17]
was used to calculate the crystallite size (
τ), where k is a constant (0.89),
λ is the X-ray wavelength,
β is the full width at half maximum (FWHM) of the diffraction line and
θ is the diffraction angle.
θ and
β were taken for (1 0 1) and (1 1 0) crystal plane of anatase and rutile phase, respectively. The contribution of size and strain to peak broadening was estimated using the Williamson–Hall (W–H) Equation (3): [
18]
where ε is the microstrain, and also with the size–strain plot (SSP) in accordance with the Equation (4): [
19]
where K’ is a particle shape dependent constant, e.g., 0.75 for spheres.
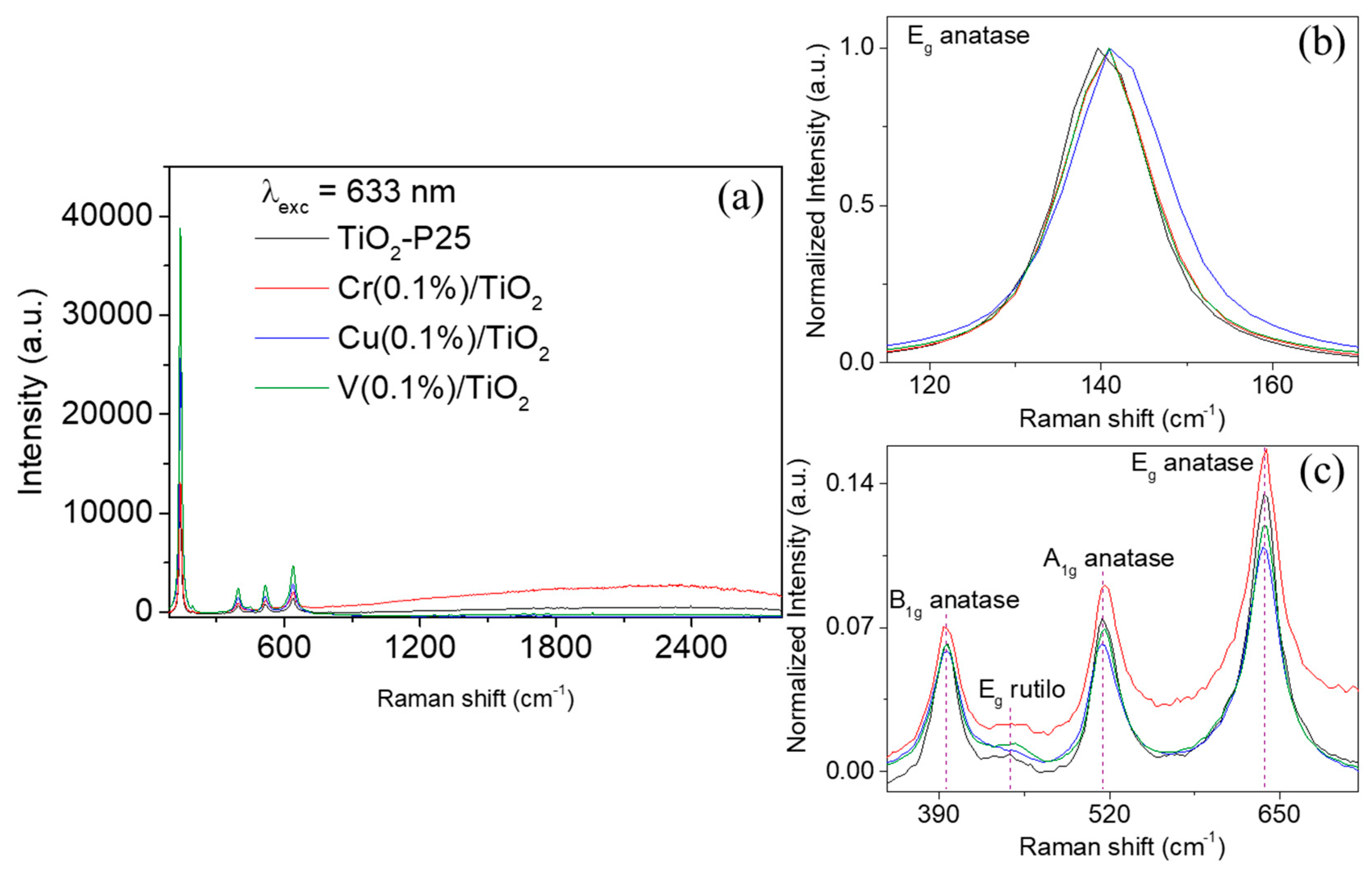
The composition of the catalyst after its use was tested by semi quantitative X-ray fluorescence on a S4 Pioneer Bruker X-ray spectrofluorometer, equipped with Rh/Ag tube and analyzer crystals LiF200, Ge, PET, OVO-8 and OVO-55. Raman measurements of the dried samples on glass were performed using a Witec Alpha 300 R confocal Raman system equipped with a 633 nm excitation laser line (10× objective), holographic 600 gr⋅mm−1 grating and Peltier-cooled CCD detector (–70 °C). Raman spectra were acquired at room temperature over a total spectra range of 90–2800 cm−1 (2.7 cm−1 spectrum resolution) with ten 2 s accumulations and laser power at the sample of 21 mW. The laser was focused onto the sample by using a 10× objective (N.A. 0.2) providing a laser spot of ca. 3.8 µm. The Raman band of a silicon wafer at 520 cm−1 was used to calibrate the spectrometer. A simple baseline (as vertical setoff; y = 0) were applied to each spectrum using Spectragryph 1.2.11. The spectra were normalized to the Eg peak (142 cm−1) for a better comparison between samples The positions and widths of the peaks were extracted by fitting the spectrum with pseudo-Voigt functions using Project FOUR (® 2014, Witec GmbH, Ulm, Germany).
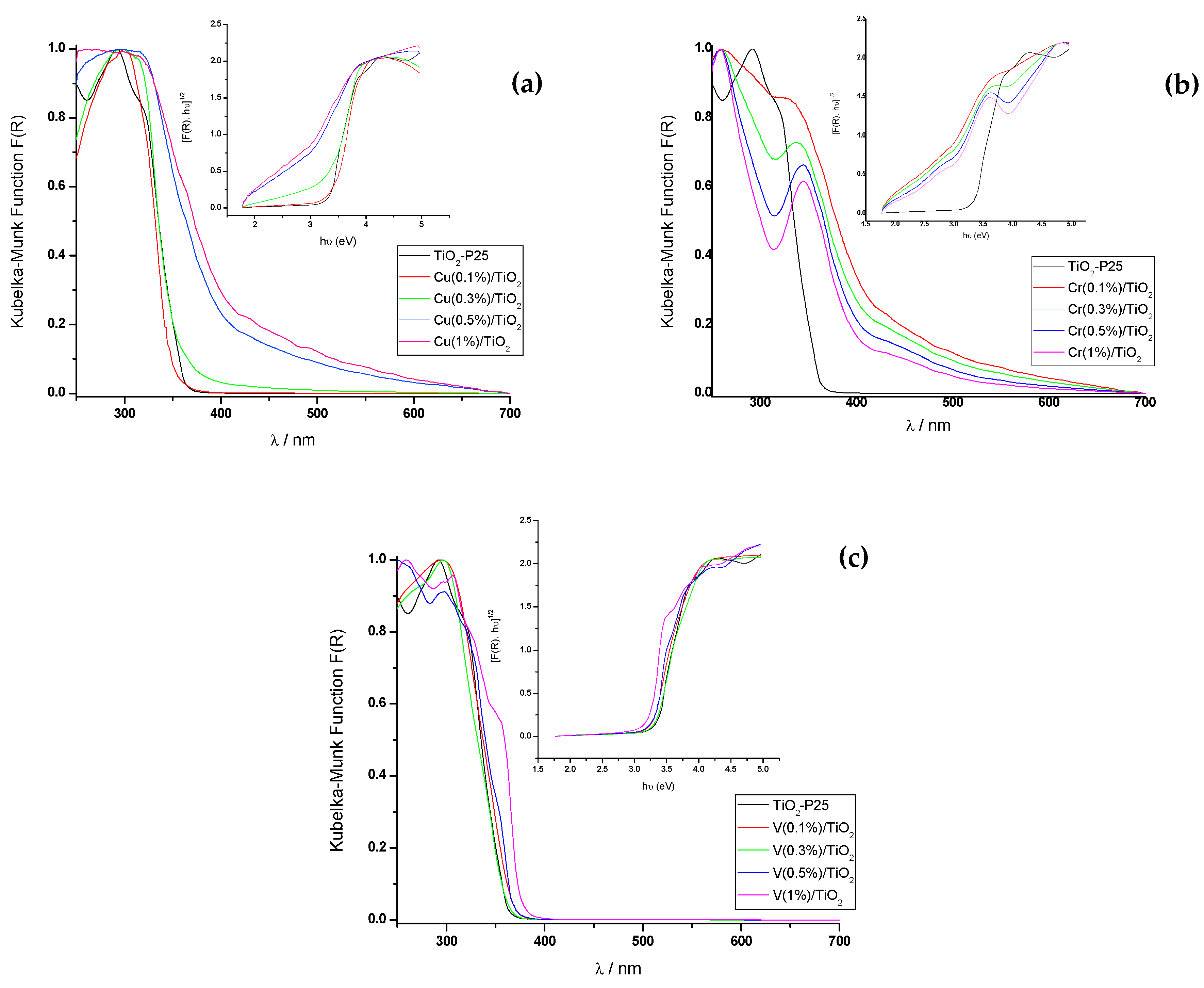
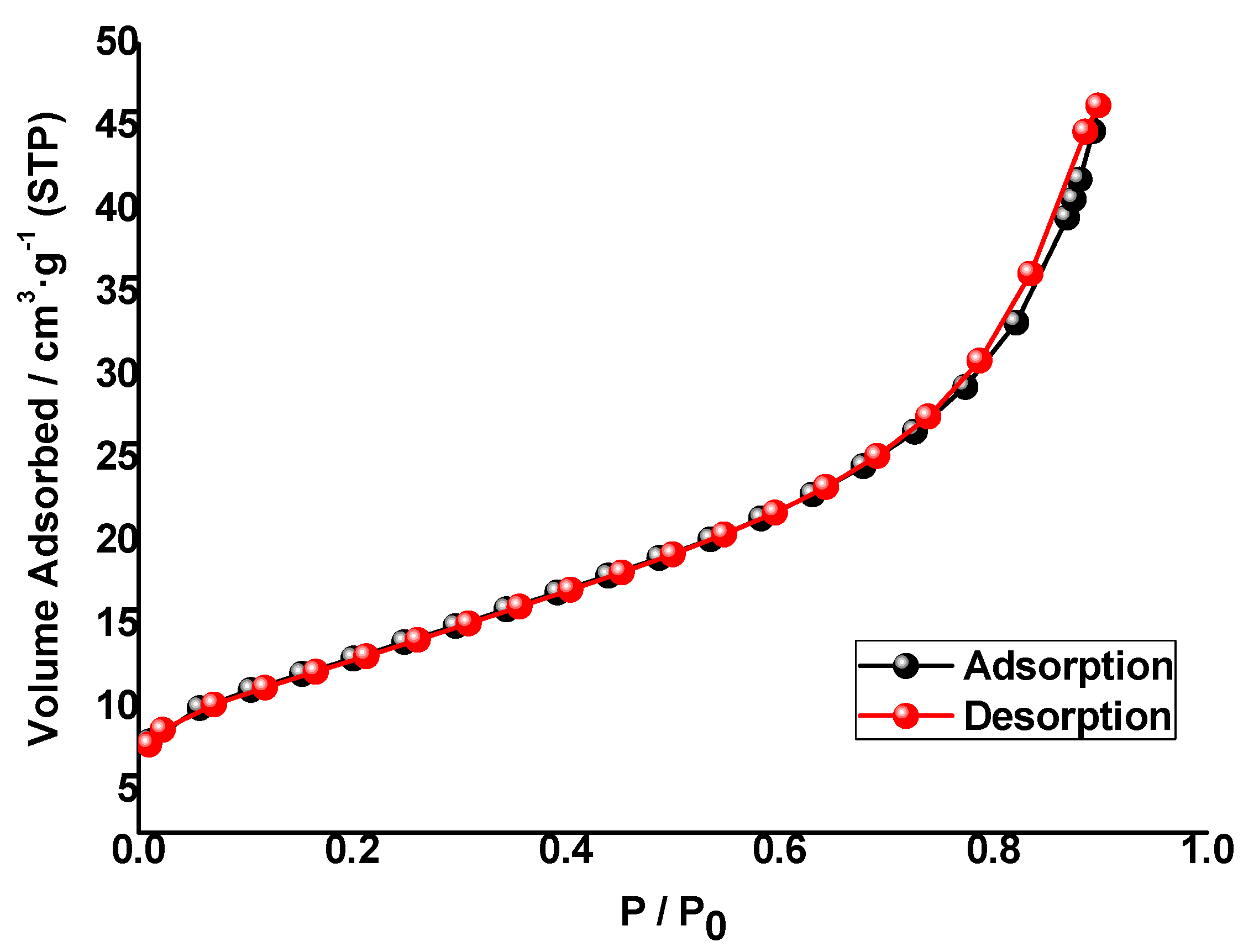
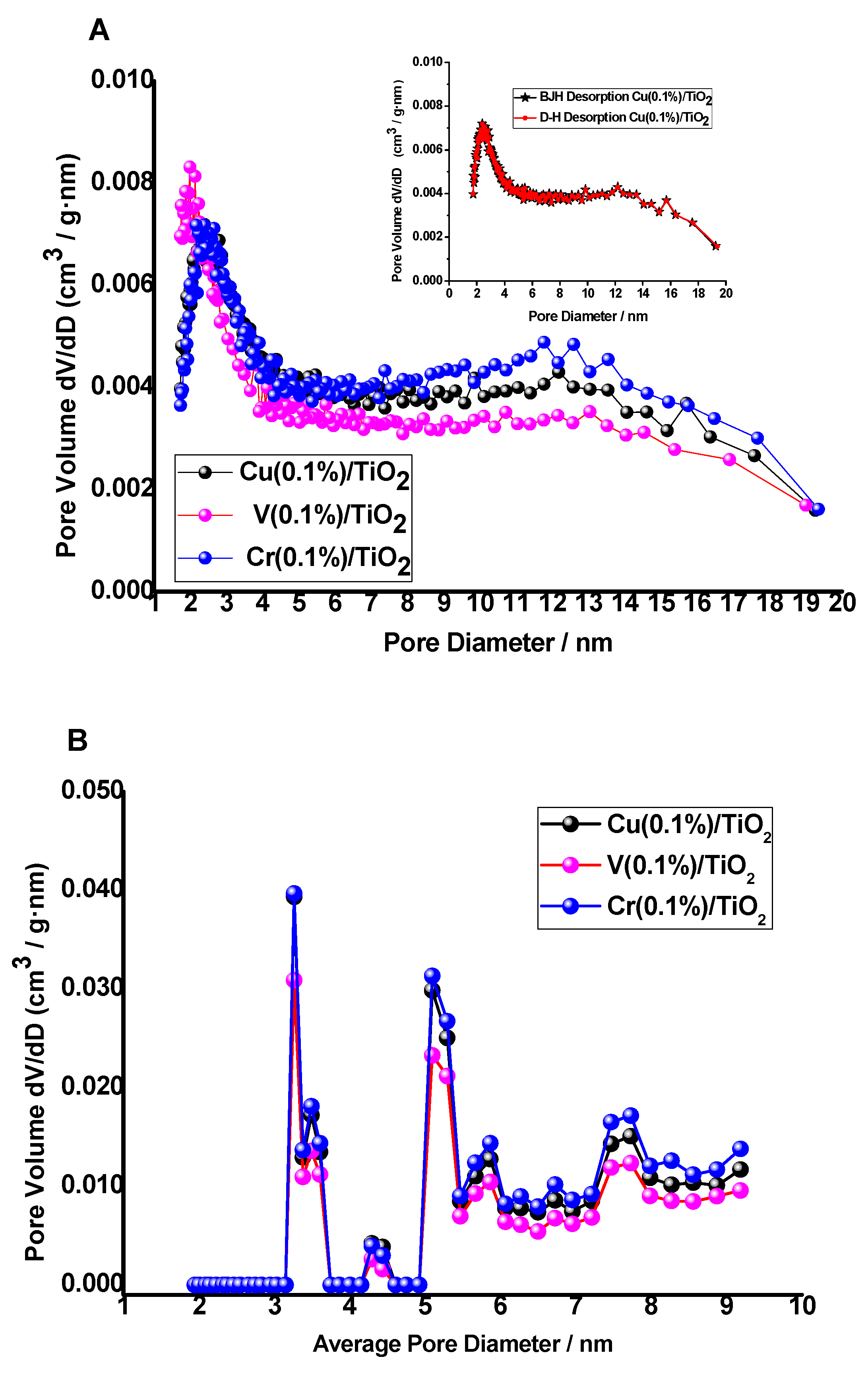
The UV-Vis diffuse reflectance (DRS) spectra (200–800 nm) of solid photocatalysts were measured on a JASCO V-560 UV-Vis spectrophotometer with a double monochromator and double beam optical system, equipped with an integrating sphere attachment (JASCO ISV-469, Oklahoma City, OK, USA). Reflectance spectra were converted by the instrument software (JASCO) to equivalent absorption Kubelka–Munk units. BET surface areas of the photocatalyst samples were measured using a BET equipment Tristar II Plus (Micromeritics; automatic station with 3 simultaneous measurement ports). The isotherms were measured in the range P/P0 = 0.1–1.0. The gases used were He (for the measurement of the dead volume of the sample holders) and N2 as adsorption gas. The measurements were made at the temperature of liquid nitrogen (77.4 K). The BET zone range used for the calculation of the specific surface area was P/P0 = 0.05–0.3. The software used for control, acquisition and data processing was “Microactive for Tristar II Plus”, v.2.03 (Micromeritics). To test the stability of the photocatalysts, the filtrate composition was analyzed by ICP-MS (Perkin-Elmer model NexION 300D).
The topography of the metal impregnated photocatalysts was calculated as roughness exponent, also known as fractal dimension D
S, using the Frenkel–Halsey–Hill Equation (5): [
20,
21,
22,
23]
where
Slg is the adsorbed amount of nitrogen at the relative pressure
P/
P0, D
S relates to solid roughness and its adsorption and permeability capacity, and μ is the adsorption potential (Equation (6)):
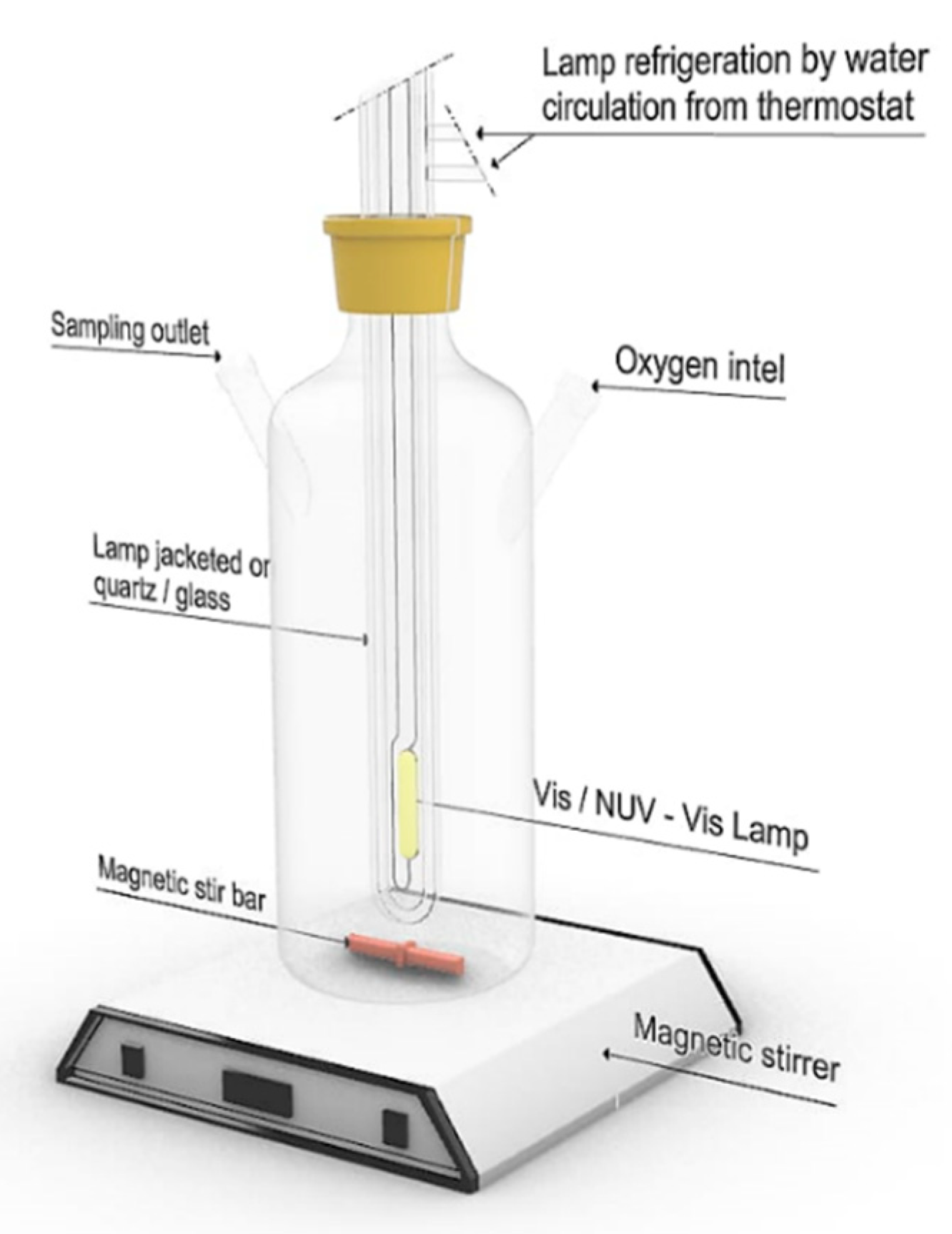
2.4. Photocatalytic Activity
The photocatalytic activity under UV and near UV-Vis light (NUV-Vis) of the synthesized photocatalysts was tested by monitoring the concentration changes of an aqueous phenol solution (C
6H
5OH) in an annular immersion photoreactor (shown schematically in
Figure 1).
Experiments under NUV-Vis irradiation were carried out with a medium-pressure Hg vapour lamp, with intense emission lines at
λexc = 254, 313, 366, 405, 436, 546, and 578 nm. UV lines at
λexc < 366 nm were filtered using a DURAN 50
® glass jacket filled with water. The photon flux at 366 nm, as determined by potassium ferrioxalate actinometry [
24], was 2.38 × 10
–6 Einstein·s
−1. Experiments under UV irradiation were carried out with a low-pressure Hg vapor lamp, with a single intense emission line at
λexc = 254 nm, located axially in the reactor inside a quartz tube. The photon flux at 254 nm, as determined by potassium ferrioxalate actinometry [
24], was of 8.33 × 10
–8 Einstein·s
−1.
Unless otherwise stated, the different reactions were carried out with 200 mL of 50 ppm phenol solutions and 200 mg of photocatalyst, in all cases in the presence of O
2, under magnetic stirring. The concentration of O
2 was routinely tested on the water used, and it was according to the expected solubility at the experimental temperature. All photocatalyst suspensions were allowed to equilibrate in the dark for 30 min as we know from our previous work that this time is sufficient to allow the establishment of the adsorption–desorption equilibrium [
25].
The different heterogeneous suspensions were irradiated for 60 min under UV light or for 300 min under NUV-Vis light. Aliquots were withdrawn at given time intervals, and filtered through Sartorius NY 0.45™ filters, for phenol and total organic carbon (TOC) analysis. All kinetic runs were performed at 298.0 K, the temperature being maintained by water flow from a thermostat–cryostat. The pH of the medium was the natural pH of the solution, given by the mixture of the photocatalyst and phenol (pH(Cu/TiO2) = 4.7, pH(V/TiO2) = 4.9, pH(Cr/TiO2) = 5.0).
The photocatalytic degradation efficiency was calculated based on the initial phenol concentration. [Phenol] was monitored by measuring the UV-Vis absorbance at 270 nm, using a Biochrom Libra S70 spectrophotometer, and by UV-Vis HPLC analysis at 210 and 270 nm, in a Thermo Fisher apparatus, equipped with a 6000 LP UV detector, an AS 3000 autosampler and a P4000 solvent pump. A Kromaphase C18 column (4.6 mm × 150 mm × 5 μm) was used, with an injected volume of 50 μL, a flow rate of 1.0 mL·min−1, at 30 °C, with acetonitrile: water (25:75, v/v) as mobile phase. The TOC removal efficiency was measured using a ShimadzuTOC-5000A analyzer.
Photoproducts were identified using HPLC/MS (Thermo Scientific LTQ Orbitrap Discovery apparatus), equipped with an electrospray interface operating in negative ion mode (ESI-). A Phenomenex Kinetex XB-C18 column (100 mm × 2.6 μm) was used, operated at 30 °C with elution solvents A (0.1% formic acid) and C (0.1% methanol.) at flow rate of 200 μL·min−1. The gradient was as follows: 0–1 min, 95–95% A and 5–5% C; 1–8 min, 95–5% A and 5–95% C; 8–10 min, 5–5% A and 95–95% C; 10–11 min, 5–95% A and 95–5% C; 11–15 min, 95–95% A and 5–5% C. Typical injection volumes were 5–25 μL. The analyses were carried out using full-scan data dependent MS scanning from m/z 50 to 500.
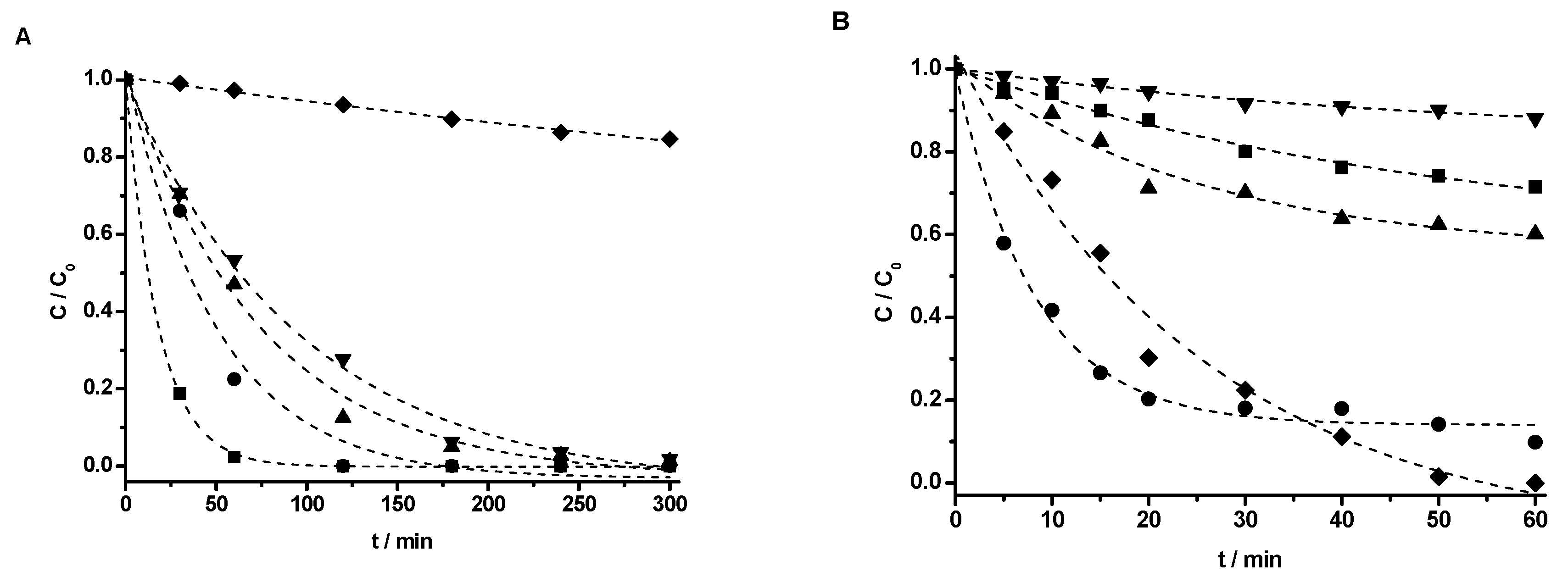
4. Photodegradation of Phenol under Vis and UV Light Irradiation
The photocatalytic activity of M(X%)/TiO
2 samples was tested by monitoring phenol degradation in aqueous solution under NUV-Vis (
λexc > 366 nm) and UV (
λexc = 254 nm) irradiation. The experimental data were in all cases adequately fit by a first order kinetic model (C = C
0·e
–k·t), and the corresponding apparent first order rate constants are collected in
Table 4. The rate constants obtained by UV-Vis spectrophotometry and HPLC, with UV detection, are comparable, and the small differences observed follow the same pattern. The effect of impregnation with transition metal ions at different concentrations on phenol removal efficiency is displayed in
Figure 8 for Cu, as a prototypical example, and all other effects are shown in
Figures S15–S24 in the Supplementary Information.
Minor or no changes were found in the kinetics and efficiencies of the process upon three repeated cycles of photodegradation using the same batches of photocatalysts.
Phenol photocatalyzed degradation under UV light irradiation is, in general, faster using non-impregnated TiO
2-P25 (
Figure 8B and
Table 4). Though for 0.5% Cu the process is faster than for non-impregnated TiO
2-P25 in the first 30 min, the efficiency of the process with 0.5% Cu is lower, not reaching full disappearance of phenol. Similar to previous studies, there is an optimum dopant concentration [
30,
54], here clearly 0.3% for Cu, 0.1% for V, and 0.5% for Cr (
Figure 8B and
Table 4). Limited phenol removal, measured as the loss of absorbance of the reacting mixture at 270 nm, was found using metal impregnated TiO
2-P25 photocatalysts under UV irradiation, usually lower than 30% (
Figure 8B and
Figures S19–S24 in the Supplementary Information).
All metal-impregnated photocatalysts showed phenol removals much faster and efficient than the standard TiO
2-P25 under NUV-Vis light irradiation.
Figure 8A shows complete phenol degradation under NUV-Vis light irradiation using TiO
2-P25 coated with Cu, much faster using Cu(0.1%)/TiO
2-P25 (half-life ca. 15 min) than Cu(1%)/TiO
2-P25 (half-life ca. 43 min). This time the lowest metal content, 0.1%, showed the best result, both in terms of rate and efficiency (understood as reaction extent after 300 min). A similar behavior was described in the photocatalyzed degradation of Malachite Green, as here the fastest photodegradation rate was found for non-impregnated TiO
2 under UV radiation, whereas it was the slowest under Vis and direct sunlight irradiation. The maximum photodegradation rate was found at Cu(1.71%)–TiO
2 [
49] under Vis irradiation. A similar behavior was also observed in the NUV-Vis photodegradation of methyl orange, maximum at Cu(1%)–TiO
2 [
63].
The photocatalytic efficiency for V/TiO
2-P25 photocatalysts, under NUV-Vis irradiation, was 99% (59%) after 175 min (300 min) for V(0.1%)/TiO
2-P25 [V(1%)/TiO
2-P25]. An optimum dopant concentration has been reported for the degradation of Methylene Blue using V doped TiO
2; [optimum dopant concentration V(0.5%)–TO
2] [
29], for 4-nitrophenol using [V(0.5%)–TiO
2 ] [
34], and for 2-4-dichlorophenol [V(1%, as V
4+)–TiO
2] [
32].
In the case of Cr, the removal yield reached the highest value of 70% for Cr(0.1%)/TiO
2 after 300 min under Vis irradiation. The photoactivity of Cr/TiO
2-P25 is similar under Vis and NUV irradiation and almost independent of the Cr content and slightly higher than with bare TiO
2-P25, such behavior matches that of previously reported [
23,
64,
65].
Here the differences in reactivity do not come from the crystallite size (
Table 1) or from the surface roughness (vide supra), they are similar for the three photocatalysts. In addition to crystalline structure and specific surface area, there are other factors playing a relevant role in the photocatalytic activity of metal impregnated photocatalysts; metal dopants might behave as hole (h
+) and/or electron (e
−) traps, therefore changing the electron/pair recombination rate.
The photocatalytic activity of metal impregnated TiO
2-P25 does not run parallel to the increase of the red-shift of absorption edge and light absorption in the visible light region with dopant concentration. It is well known that beyond an optimum dopant concentration, here M(0.1%)/TiO
2-P25, the photocatalytic activity decreases [
30]. Several reasons can be argued to explain such photoactivity decline after the dopant concentration optimum. First, it might be due to the increase of e
−/h
+ pair recombination as dopant concentration increases. A higher concentration shortens the distance (R) between trap sites of photogenerated e
−/h
+ pairs and the recombination rate (k
RR) of charge carriers increases in accordance with the equation:
where a
0 is the radius of the hydrogenic wave function for the charge carrier, i.e., metal dopants become recombination centers as the distance between trapping sites shortens [
46]. The low photocatalytic effectiveness of Cr-impregnated TiO
2-P25 likely due to the short diffusion length of the charge carriers (ca. 0.2 μm) [
65], leading to a faster e
−/h
+ recombination rate [
32,
63].
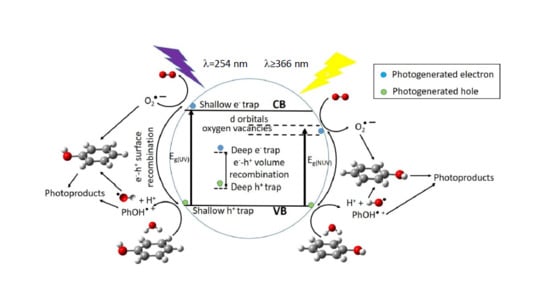
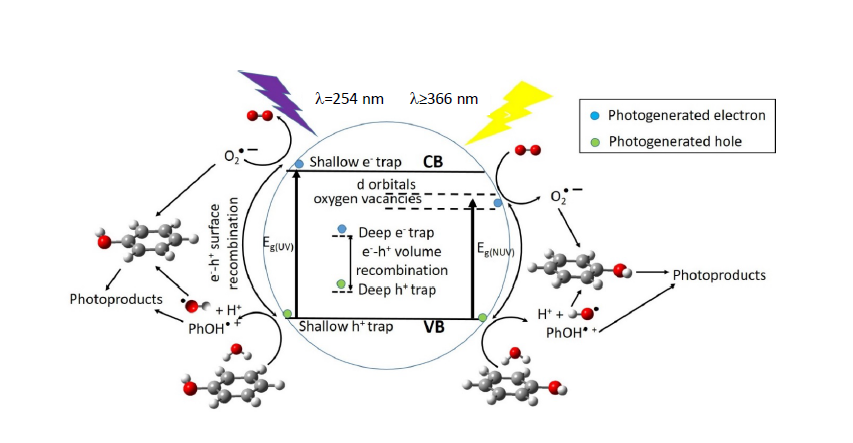
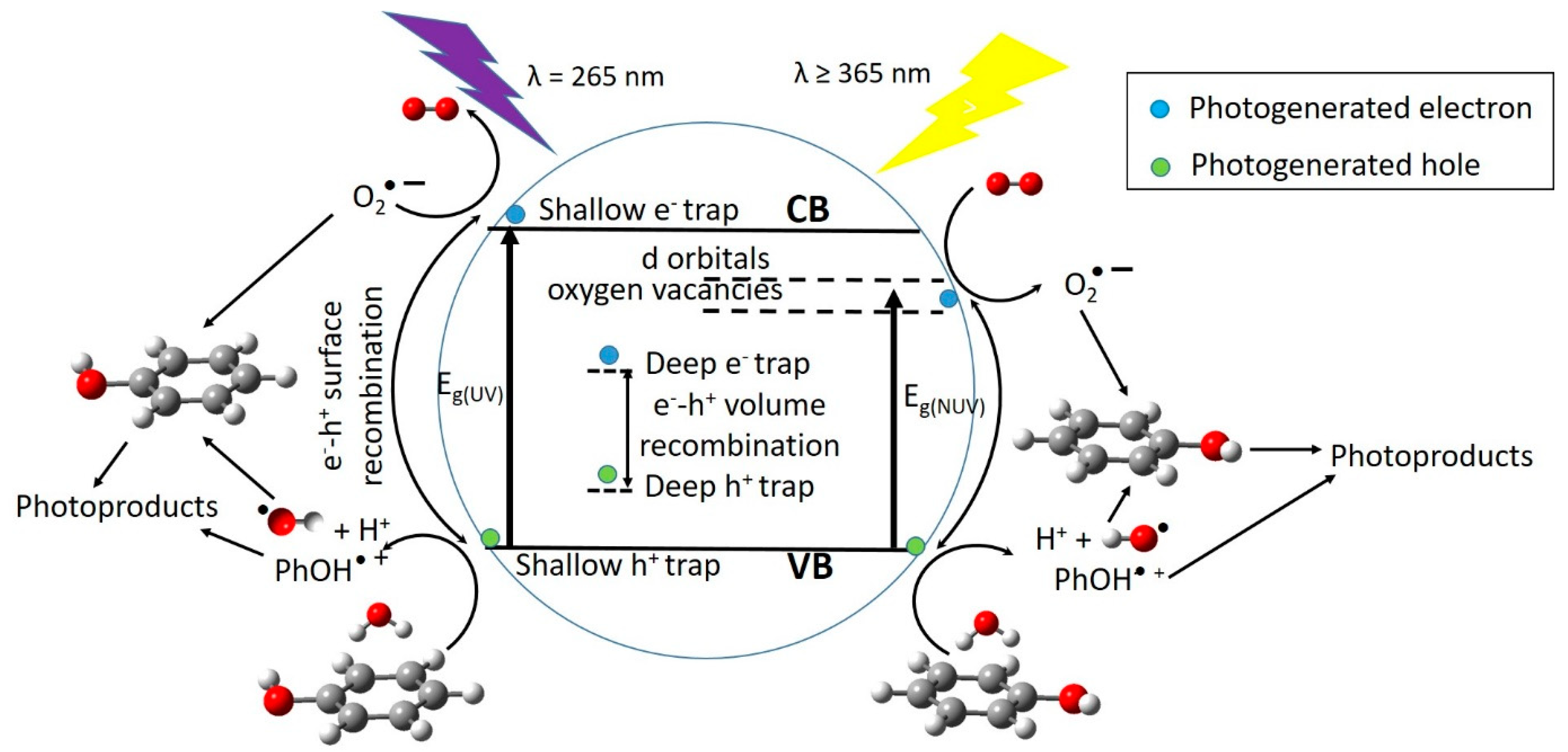
At the optimum surface impregnation concentration there is an efficient separation between photogenerated h+ and e−. The space charge region extends and the surface barrier for recombination is maximum, whereas at higher dopant concentration the space charge region narrows, e−/h+ pairs are produced in the bulk of the photocatalyst, deep trap instead of shallow trap takes place, and volume recombination dominates leading to reduced photocatalytic activity.
Second, a decrease in specific area with concentration means reduction of photoactivity. Third, we hypothesized that the shading effect due to the higher surface coverage as the dopand concentration increases, which also means minor contact area between phenol and TiO
2-P25 [
32]. Fourth, the metal ion can bind to surface hydroxyl groups thus reducing the availability of –OH groups to be converted into HO radicals. The increased photocatalytic activity of Cu and V-impregnated TiO
2-P25 in the degradation of phenol, under NUV-Vis light irradiation, points to different mechanism of photoactive enhancement under UV and NUV-Vis radiation. In the latter, photoelectrons are transferred from impregnated TiO
2-P25 VB to Cu or V 3d-orbitals lying just below of the CB, then migrate to form O
2– radicals, whereas holes migrate to the surface, react with HO
– rendering HO radicals, then the so-formed radicals are able to initiate the degradation of the adsorbed phenol molecules (
Figure 9). Metal ions with charge different than Ti
4+ could produce oxygen vacancies in the lattice with energy levels below the TiO
2-P25 CB (
Figure 9), allowing visible light harvesting, acting as active sites for adsorbed water dissociation and capturing holes to diminish electron-hole recombination, thus enhancing the photocatalytic activity [
35,
66,
67].
Photocatalytic activity, among other factors, is not only dependent on the photogenerated charge carriers trapping, efficient detrap to the surface should also occur. Metal dopants can act as h
+ traps (M
n+ + h
VB+ → M
(n+1)+) and/or e
− traps (M
n+ + e
CB– → M
(n–1)+), the energy levels of M
n+/M
(n+1)+ and M
n+/M
(n–1)+ lying above VB and below CB of TiO
2-P25, respectively [
47]. The stability of M
(n+1)+ and M
(n–1)+ depends on the change on the electronic configuration relative to the initial electronic configuration of M
n+, for instance e
− trap, better than h
+ trap should occur for Cu
2+, in this way d-orbitals become completely filled. V
5+ and Cr
3+ should act as hole traps, whereas, for example, it is accepted that V
4+ serve both as h
+ and e
− trap [
47]. Then M
(n–1)+ can transfer the trapped electron to the TiO
2-P25 lattice, and from there to adsorbed O
2 molecules to yield O
2–, or trap a VB h
+ turning back to its stable electronic configuration. On the other hand, M
(n+1)+ can transfer its additional positive charge either to adsorbed HO
–, forming the reactive HO radical, or to adsorbed phenol molecules (
Figure 9).
In summary, all the studied impregnated photocatalysts were less efficient under UV irradiation (extent of reaction after 60 min) than the standard TiO2-P25, where photoactivity follows the order: TiO2-P25 >> Cu > V > Cr, irrespective of the metal percentage in the photocatalyst; whereas under NUV-Vis irradiation, phenol photocatalyzed degradation over M(%)/TiO2 composites is faster and more efficient, irrespective of the dopant content, than with TiO2-P25, the reactivity order being Cu > V >> Cr > TiO2-P25 (extent of reaction after 300 min).
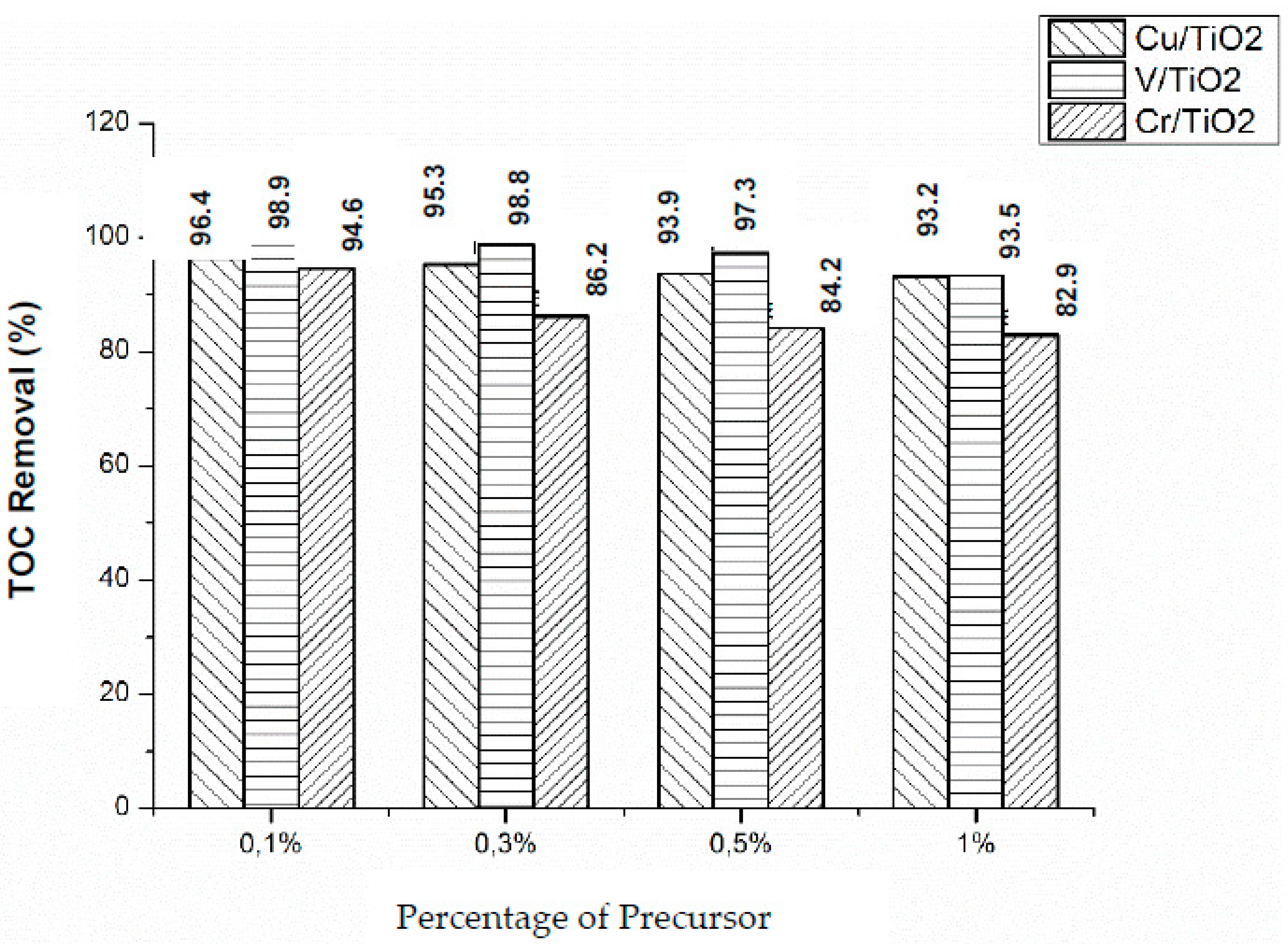
Total Organic Carbon
TOC measurements were carried out to determine the degree of mineralization reached, under both Vis and UV light irradiation.
Figure 10 shows the obtained results for the three photocatalysts (X = 0.1%, 0.3%, 0.5%, and 1%) under Vis irradiation. V/TiO
2 was the most efficient photocatalyst for TOC removal, with the maximum removal observed for the lowest metal content (0.1%), decreasing as the metal percentage increases, in accordance with kinetic results. On the other hand, Cr/TiO
2 photocatalysts are the less efficient in terms of TOC removal. Comparison between kinetic observations and TOC results suggests that phenol disappearance is faster than mineralization, some organic intermediate photoproducts remain in solution, and the amount depends on the impregnation metal and its concentration. Similar behavior was observed with UV light irradiation (
Figure 11), although much lower TOC removals were obtained with the three metal impregnated photocatalysts, which is consistent with kinetic runs (vide supra). The most efficient, in terms of TOC removal, was Cu(0.1%)/TiO
2.
Phenol photocatalyzed degradation over M/TiO
2 (M: Cu, Cr, and V), measured either as phenol disappearance or as TOC removal, is higher under NUV-Vis light than under UV irradiation. We have previously found very high efficiencies of NUV-Vis photocatalytic TOC removal using photocatalysts doped with Cu [
25]. Thinking in pollution abatement, although slower, photodegradation is more effective under NUV-Vis, and in economic terms longer time using NUV-Vis radiation counterweight the costs of using UV irradiation sources.
5. Reaction Pathways for Photocatalyzed Degradation
Photocatalytic degradation of phenol is a complex multi-stage process. The photocatalytic process with M–TiO
2 (M = Cu, V, and Cr) is energetically favourable for the decomposition of phenolic compounds with respect to the process with the standard TiO
2–P25. Two types of oxidizing species, i.e., the radical hydroxyl HO and superoxide O
2– are involved in the transformation of the aromatic compounds [
15]. The lifetime of the intermediates formed at different stages of the reaction is short, as they undergo further fast catalytic oxidation.
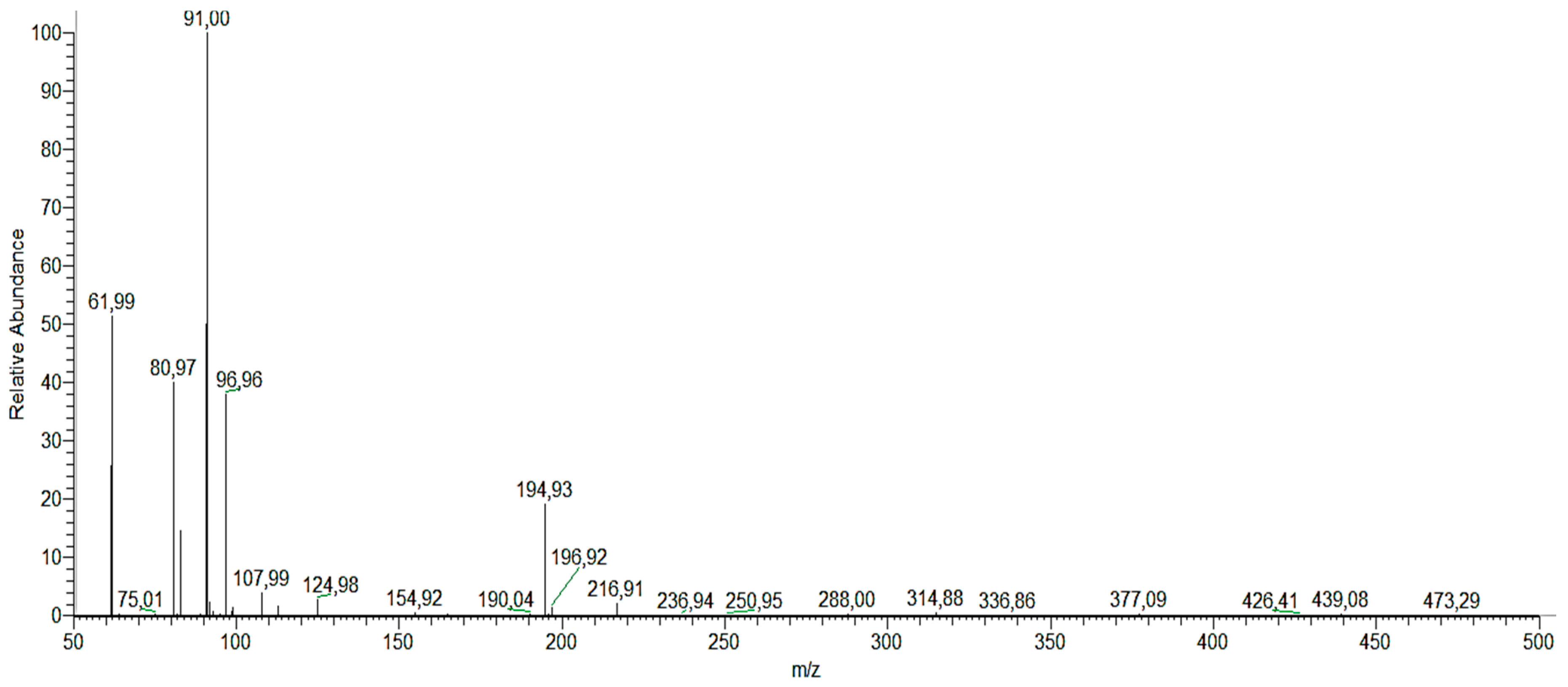
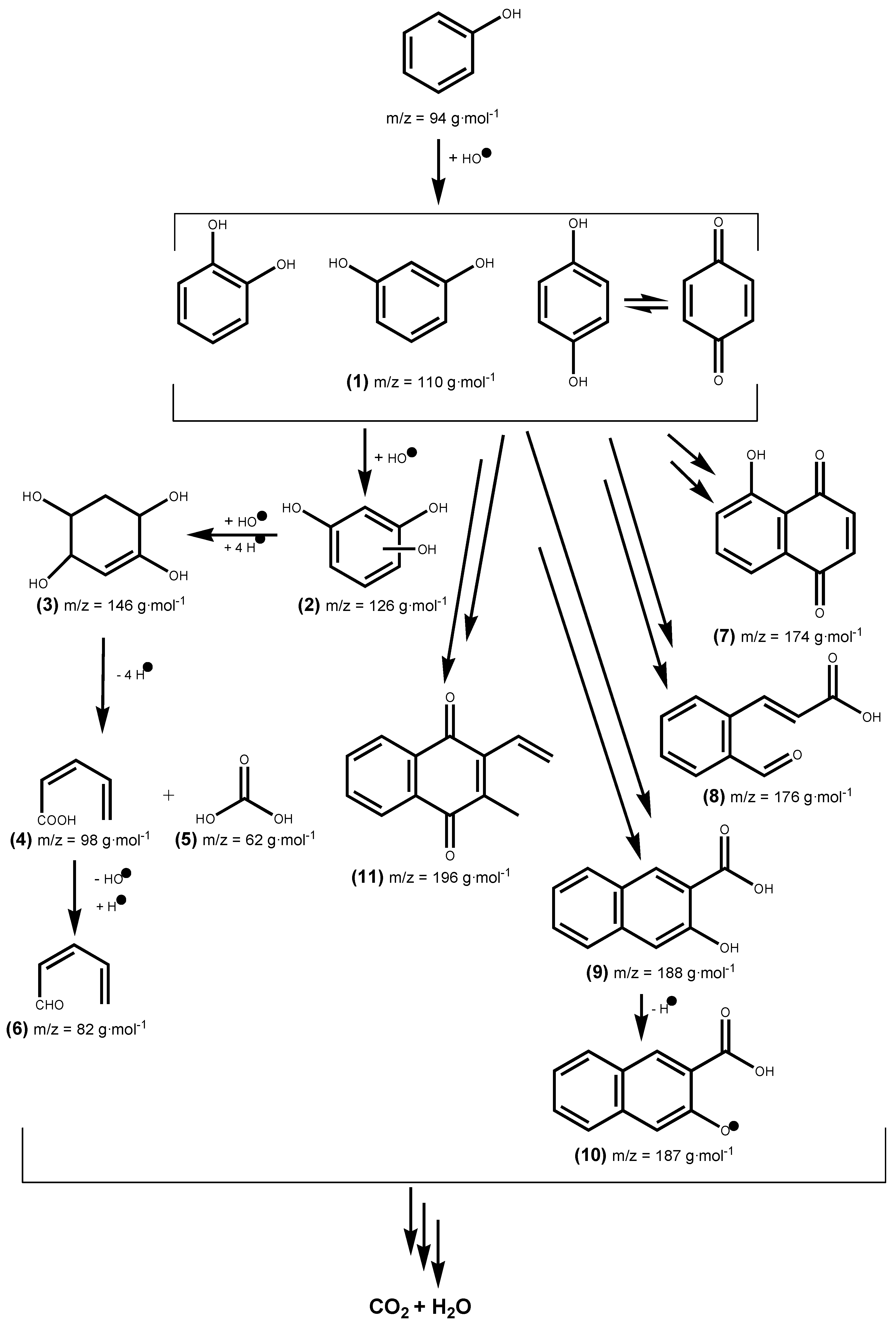
We determined the different reaction intermediates using HPLC–MS.
Figure 12 shows a typical HPLC chromatogram. The photoproducts found under both UV and Vis light are summarized in
Table 5. Identification of the intermediate products is based on the obtained MS and compared to those in databases. The main intermediates identified in this way were:
(1) catechol, resorcinol and/or hydroquinone,
(2) phloroglucinol,
(3) cyclohex-2-ene-1, 2, 4, 5-tetraol,
(4) (Z)-penta-2,4-dienoic acid,
(5) carbonic acid,
(6) (Z)-penta-2,4-dienal,
(7) juglone,
(8) 9H-xanthen-9-one,
(9) 3-hydroxy-2-naphthoic acid,
(10) 3-Hydroxy-2-naphthoate, and
(11) (2E)-3-(2-formylphenyl) acrylic acid. Degradation pathways into smaller molecules are proposed and also routes leading to heavier transformation products.
Phenol photocatalyzed degradation over titania-coated metal composites under UV and Vis light irradiation is described by the reaction mechanism shown in
Figure 13. Photocatalyzed phenol degradation proceeds through the widely accepted mechanisms of electrophilic attack promoted by HO or h
+ oxidation of the adsorbed phenol onto the photocatalyst surface [
66]. Though a deep understanding of the surface processes taking place is out of the scope of this article, in support to the previous statement, we have observed that the presence of
iso-Propanol as HO scavenger or EDTA as h
+ scavengers inhibit the process largely or completely.
Phenol oxidation occurs by hydroxylation to yield dihydroxylbenzenes (
1) (catechol, resorcinol, and/or hydroquinone) [
67,
68]. Further hydroxylation produces phloroglucinol (
2) and likely other trihydroxybenzenes. The hydroxylation of the former with addition of hydrogen gives (
3) which leads to intermediates (
4) and (
5) by ring opening via C–C bond breaking, then dehydroxylation of (
4) and hydrogen addition leads to (
6). Several radical species are formed in phenol photodegradation [
69,
70,
71,
72] that can react between them to give the other intermediates (
7–
11) found in this work.
6. Photodegradation and Energetic Efficiency of the Process
Light scattering by the suspended catalyst particles and the characteristics of the surface contribute to reduce the photodegradation quantum yield (Φ
photodegradation) and the photonic efficiency of the process (ξ) [
73].
The photocatalysis quantum yield (Φ
Photocatalysis) is the ratio of moles of reactant consumed per Einstein absorbed by the photocatalyst [
74]. It can be calculated as [
75,
76,
77]:
where Φ
photodegradation is the photodegradation quantum yield, k
app is the apparent pseudo first order rate constant,
Iλ (Einstein·L
−1·s
−1) is the light intensity at wavelength
λ, ε
λ (cm
−1·mol·dm
–3) is the molar absorptivity at
λ, and l is the path length of the photoreactor (cm). The so-obtained Φ
photodegradation are shown in
Table 6. Both for UV and UVA-Vis lamps, Φ
photodegradation (Cu) > Φ
photodegradation (V) > Φ
photodegradation (Cr), with values higher than 1 for Cu and V, pointing to the existence of secondary processes, that inflate the efficiency of the incident photons.
Photodegradation of organic micropollutants in aqueous solution is energy demanding, and an energy efficiency parameter (E
EO) can be defined to analyse it, as the kWh of energy required to reduce the pollutant concentration per volume and time unit (kW·L
−1·s
−1) [
78], expressed as:
where P is the electric power consumed by the lamp (kW), V is the volume (L) of solution and k
app are the apparent photodegradation rate constants from
Table 4. The values thus obtained for E
EO, compiled in
Table 6, are lower with 254 nm than with 366 nm, showing that E
EO is far more favourable with UV lamps. The efficiency order varies, both for UV and UVA-Vis lamps, in the order E
EO (Cu) < E
EO (V) < E
EO (Cr).
7. Conclusions
The photocatalyzed degradation of phenol, as a model pollutant, in aqueous solution over titania-coated metal (M = Cu, Cr, and V) composites under visible (λexc > 366 nm) and UV (λexc = 255 nm) irradiation is described. Metal surface impregnated photocatalyts were synthesized using the wet impregnation method and characterized by X-ray diffraction (XRD), X-ray fluorescence (XRF), UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS), and surface area (BET). No oxides phases of the metal dopants were found. Analysis of XRD peak broadening, in terms of the uniform deformation model (UDM), points to the existence of tensile strain in Cu-impregnated TiO2-P25, whereas compressive in Cr- and V-impregnated TiO2-P25.
UV-Vis DRS measurements clearly show the dependence of the band gap on the synthetic method. Raman spectra showed an enhancement of the crystallinity of the anatase phase by impregnation mainly with Cu and V cations. Red-shifts were also observed on the Eg (140 cm−1) peak (ΔRaman-shift = 2.8–4.4 cm−1) in M(0.1%)/TiO2 samples, especially for Cu(0.1%)/TiO2. Experimental evidences suggest at least a surface metal-linkage.
Type IV N2 adsorption–desorption isotherms were found, with a small H3 loop near the maximum relative pressure. SBET was similar for the three M(0.1%)/TiO2 photocatalysts, ca. 45 m2·g−1. Pore size distribution using BJH, D–H, and NLDFT models suggest those photocatalysts are mesoporous with a narrow pore distribution centered at ca. 25 Å, which is in agreement with the rest of textural data. PSDs using BJH and D–H models are fully coincident, and the same is observed using adsorption and desorption branches. Metal surface impregnated photocatalysts show similar surface roughness.
Their photocatalytic activity was followed by UV-Vis spectroscopy and HPLC–UV. A first-order equation was used to fit kinetic data. Similar results are obtained using UV-Vis spectroscopy and HPLC monitoring. Fastest phenol degradation was obtained with M(0.1%)/TiO2 the order being Cu > V >> Cr > TiO2/P25 under Vis radiation, whereas reactivity was TiO2/P25 >> Cu > V > Cr under UV radiation.
The degree of mineralization was calculated in terms of TOC removal efficiency, and we were able to achieve more than 95% disappearance of the total organic carbon using visible light and less than 30% when treated with UV light.
Products and intermediate organic photoproducts were identified by HPLC–MS spectrometry, and the corresponding kinetic mechanism proposed.
Finally, the energetic efficiency of the process was analysed for M (0.1%), showing that UV lamps are far superior to UVA lamps, and that the efficiency of the surface impregnated catalyst varies in the order EEO (Cu) < EEO (V) < EEO (Cr).

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}